I. INTRODUCTORY REMARKS
Most of the previous chapters of this series have emphasized gross organization and structure of the brain. This has been essential in order to gain perspective, but from a cryonicist's point of view preservation of the "the anatomical basis of mind" will ultimately mean preservation of the structures only visible under a microscope. Understanding what structures to look-for and how those structures might best be preserved is the ultimate goal of this series.
As a step in the direction towards understanding finer structure, this chapter will examine the brain from a more chemical point of view than the previous installments — with particular reference to the gross anatomy and function of neurotransmitters in the brain.
II. THE CHEMICAL ENVIRONMENT OF THE BRAIN
![[Skull surface dissection]](./FigX1.gif)
|
The brain has the
consistency of firm
jelly, and therefore is
protectively encased in
a thick, bony skull.
The brain literally
floats in about 150
millilitres (mL) of
CerebroSpinal Fluid
(CSF) secreted by the
choroid plexus.
Approximately 500 mL
of CSF is secreted daily,
which slowly circulates
down through the four
ventricles, up through the
subarachnoid space and exits
into the cerebral veins
through the arachnoid villi.
The brain has no lymphatic
system, so the CSF serves
as a partial substitute.
![[Skull section]](./FigX2.gif)
|
![[Brain ventricles]](./FigX3.gif)
|
The dura mater is a
tough, protective connective tissue which is
tightly bound to the skull,
but which encases the
cerebral veins. Under
the dura mater is the
subarachnoid space containing CSF, arteries and
web-like strands of connective/supportive tissue called the
arachnoid ("spider-like")
mater. The pia mater is a
permeable membrane of collagen, elastin fibers & fibroblasts on the
floor of the subarachnoid
space which allows diffusion
between the CSF and the
interstitial fluid of the
brain tissue. The pia mater lies on a membrane that is infiltrated with
astrocyte processes. The dura mater,
the arachnoid mater and the
pia mater are collectively
referred-to as the meninges.
![[Skull Medial Cross-Section showing CSF flow]](./FigX5.gif)
|
![[Blood-Brain Barrier]](./FigX4.gif)
|
While the brain & CSF are separated by the somewhat permeable pia mater, the blood-cerebrospinal fluid barrier and the blood-brain barrier (BBB) represent substantial protection for the brain against undesirable blood substances. These barriers are very permeable to water, oxygen, carbon dioxide and small lipid-soluble substances. They are also somewhat permeable to small electrolytes — and special transport systems exist for some other specific molecules such as essential amino acids. The barriers are the result of endothelial cells which line capillary walls — and glial cells called astrocytes which wrap the capillaries with fibers.
The brain is not only a functionally distinct organ, it is a chemically
distinct one. 50% of dry brain weight is lipid (in contrast to 6-20% for
other organs). Most of the brain lipid is structural (in myelin or membranes)
in contrast to the triglycerides and free fatty acids constituting the fat of
other organs. The blood-brain barrier creates a protected chemical environment
for the brain wherein certain molecules can perform functions independent
of the functions those molecules perform in the rest of the body. This is
particularly important for the neurotransmitters serotonin (which is
highly concentrated in platelets & the intestine) and norepinephrine
(which affects blood pressure & metabolism). All of the known amino-acid
neurotransmitters are non-essential amino acids. This means that they
can be manufactured in the brain, without needing to be supplied from
outside the brain. But in
the major area of the brain which does not have a blood-brain barrier —
the hypothalamus — the primary neurotransmitters are peptides.
III. GENERAL COMMENTS ABOUT NEUROTRANSMITTERS
The three major categories of substances that act as neurotransmitters are (1) amino acids (primarily glutamic acid, GABA, aspartic acid & glycine), (2) peptides (vasopressin, somatostatin, neurotensin, etc.) and (3) monoamines (norepinephrine, dopamine & serotonin) plus acetylcholine. The major "workhorse" neurotransmitters of the brain are glutamic acid (=glutamate) and GABA. The monoamines & acetylcholine perform specialized modulating functions, often confined to specific structures. The peptides perform specialized functions in the hypothalamus or act as co-factors elsewhere in the brain. [For a well-organized categorization of neurotransmitters, see Neurotransmitter (Wikipedia).]
Although there are many neurotransmitters in the central nervous system, the peripheral nervous system has only two: acetylcholine and norepinephrine. Why are there so many brain neurotransmitters? Because the functions performed by brain neurotransmitters are not as uniform as they might superficially appear. Some (like glutamate) are excitatory, whereas others (like GABA) are primarily inhibitory. In many cases (as with dopamine) it is the receptor which determines whether the transmitter is excitatory or inhibitory. Receptors can also determine whether a transmitter acts rapidly by direct action on an ion channel (eg, nicotinic acetylcholine receptors) or slowly, by a second-messenger system that allows for synaptic plasticity (eg, muscarinic acetylcholine receptors). Speed & mechanism of transmitter inactivation after the signal has been sent is also a factor. There are probably also costs & benefits involved in synthesizing, transporting and recycling various neurotranmitters in the differing chemical mileus of the brain.
Many of these issues will become more clear in discussing the synthesis, distribution and function of the major brain neurotransmitters.
IV. GLYCINE
![[General Amino Acid]](./FigX7.gif)
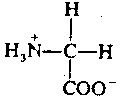
|
Glycine is the simplest of amino acids, consisting of an amino group and a carboxyl (acidic) group attached to a carbon atom. Glycine's function as a neurotransmitter is also fairly simple. When released into a synapse, glycine binds to a receptor which makes the post-synaptic membrane more permeable to Cl- ion. This hyperpolarizes the membrane, making it less likely to depolarize. Thus, glycine is an inhibitory neurotransmitter. It is de-activated in the synapse by a simple process of reabsorption by active transport back into the pre-synaptic membrane.
Glycine is a neurotransmitter only in vertebrate animals. The glycine receptor is primarily found in the ventral spinal cord. Strychnine is a glycine antagonist which can bind to the glycine receptor without opening the chloride ion-channel (ie, it inhibits inhibition). The resultant spinal hyperexcitability is what makes strychnine a poison. Quoting from the ENCYCLOPEDIA BRITANNICA:
"Symptoms of poisoning usually appear within 20 minutes, starting with stiffness at the back of the neck, twitching of the muscles, and a feeling of impending suffocation. The patient is then seized with violent tetanic convulsions in which the body is arched and the head bent backward. After a minute the muscles relax, and the patient sinks back exhausted, heightened perceptiveness being perceived throughout due to sensory cortex stimulation. A touch, a noise or some other stimulus causes the convulsions to recur; or they may recur spontaneously, often at intervals of a few minutes. Strychnine poisoning is ultimately the result of suffocation or exhaustion."
V. ASPARTIC ACID (ASPARTATE)
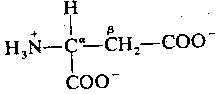
|
Like glycine, apartate is primarily localized to the ventral spinal cord. Like glycine, aspartate opens an ion-channel and is inactivated by reabsorption into the pre-synaptic membrane. Unlike glycine, however, apartate is an excitatory neurotransmitter, which increases the likelihood of depolarization in the postsynaptic membrane. Aspartate & glycine form an excitatory/inhibitory pair in the ventral spinal cord comparable to the excitatory/inhibitory pair formed by glutamate & GABA in the brain. Interestingly, the two exitatory amino acids — glutamic acid & aspartic acid — are the two acidic amino acids found in proteins, insofar as both have two carboxyl groups rather than one.
VI. GLUTAMIC ACID (GLUTAMATE)
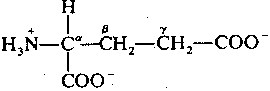
|
Glutamate is the most common neurotransmitter in the brain. It is always excitatory, usually due to simple receptors that increase the flow of positive ions by opening ion-channels. Glutamate stimulation is terminated by a (chloride-independent) membrane transport system that is only used for re-absorbing glutamate & aspartate across the pre-synaptic membrane. Glutamate & aspartate re-enter the cell by a transporter driven by the high extracellular concentrations of Na+ and the high intracellular concentrations of K+. Soduim enters the cell along with the amino acids and potassium leaves the cell — much the way a pulley couples the lifting of a light weight with the fall of a heavier weight. Thus, glutamate/asparate entry is indirectly powered by the ATP-driven Na+-K+-ase (sodium pump) which creates the high ion concentration gradients.
Possibly the most complicated of all neurotransmitter receptors is the NMDA glutamate receptor. N-Methyl-D-Aspartate is a synthetic chemical not naturally found in biological systems, but it binds specifically to the NMDA glutamate receptor (receptors are frequently named for artificial substances that bind to the receptor with higher specificity than their natural neurotransmitter ligands). The NMDA receptor is the only known receptor which is regulated both by a ligand (glutamate) and by voltage. There are at least 5 binding sites which regulate NMDA receptor activity, ie, sites for (1) glutamate (2) glycine (3) magnesium (4) zinc and (5) a site that binds the hallucinogenic substance phencyclidine (PCP, "angel dust"). Phencyclidine can induce psychosis — an NMDA effect that is difficult to explain. NMDA receptors have a capacity for an activity-dependent increase in synaptic efficiency known as LTP (Long-Term Potentiation), which may be crucial to some forms of learning & memory. Inhibition of NMDA activity (and LTP) is believed to be an important part of the way ethanol affects brain functions.
NMDA receptors are most densely concentrated in the cerebral cortex (hippocampus, especially — particularly the CA1 region), amygdala, & basal ganglia. They are particularly vulnerable to glutamic acid excitotoxicity, ie, damaging effects due to excessive excitatory neurotransmitter release. Both aspartic acid & glutamic acid (the two amino acids having 2 carboxyl groups — the "acidic amino acids") have the capacity for destroying neurons when released in excessive amounts (although calcium seems to be more of a cause than acidity). Monosodium glutamate (MSG), a major component of soya sauce, has been shown to destroy nerve cells when fed to young animals. Insofar as glutamate does not normally cross the blood-brain barrier, it is open to question whether this is relevant to a human adult. Increased alertness (or anxiety) due to caffeine may be mainly due to blockage of adenosine receptors which normally inhibit glutamate release.
Glutamate released into synapses is either reabsorbed directly into neurons by the ion-exchange transport system described above, or is soaked-up by astrocytes (glial cells) which convert the glutamate into glutamine (a molecule which cannot cause excitotoxicity). The glutamine can then be safely transported back to neurons for re-conversion into glutamate. One of the damaging effects of mercury poisoning is swelling of astrocytes, which are rendered unable to soak-up glutamine from synapses (contributing to excitotoxicity). Excitotoxicity due to glutamic acid is a major destructive process seen in stokes and other forms of brain ischemia (see Ischemia and Reperfusion Injury in Cryonics).
Nitric oxide can act as neuromodulator when glutamate stimulation of NMDA receptors results in nitric oxide synthesis & release — enhancing neurotransmitter release from adjacent synapses. Granule cells of the dentate gyrus of the hippocampus are rich in nitric oxide synthetase. Nitric oxide may contribute to LTP.
VII. GAMMA AMINO BUTYRIC ACID (GABA)
![[GABA Biochemistry]](./FigX10.gif)
|
GABA is the major inhibitory neurotransmitter of the brain, occurring in 30-40% of all synapses (second only to glutamate as a major brain neurotransmitter). It is most highly concentrated in the substantia nigra & globus pallidus nuclei of the basal ganglia, followed by the hypothalamus, the periaqueductal grey matter ("central grey") and the hippocampus. The GABA concentration in the brain is 200-1000 times greater than that of the monoamines or acetylcholine.
GABA is somewhat unique among neurotransmitters insofar as it is commonly inactivated (after release into the synapse) by active transport into the astrocyte glial cells that are closely associated with synapses. Both glutamate and GABA are synthesized in the brain from the Krebs citric acid molecule alpha-keto glutarate — a reaction known as the "GABA shunt". GABA is synthesized from glutamic acid and is catabolized back into the citric acid cycle. The vitamin B6 derivative pyridoxal phosphate is a cofactor in the synthesis of GABA, which is why seizures occur in Vitamin B6 deficiency. GABA levels rise when the citric acid cycle activity is low (ie, when cell energy usage is low), and the resultant generalized GABA inhibitory effect on the brain neurons can be protective during hypoxia or ischemia.
Like glycine, the GABA receptor is connected to a chloride ion channel, allowing more chloride ion to enter the cell and thus making the membrane less likely to depolarize. A closely associated receptor site will bind to benzodiazepines (such as diazepam) to increase the frequency of channel opening. Caffeine can neutralize the effects of benzodiazepine tranquilizers such as diazepam (Valium®). Benzodiazepines act by enhancing the effect of GABA on GABAA receptors, whereas caffeine has an opposite effect by inhibiting GABA release. Barbiturates slightly decrease the frequency of opening, but prolong the duration. The benzodiazepine receptor site is thought to be the natural site of action of a yet-unidentified peptide. By potentiating the effects of GABA, the benzodiazepines function as so-called "minor tranquilizers" (to be distinguished from the anti-psychotic "major tranquilizers"). Anxiety is the most frequently diagnosed psychiatric disorder — affecting 10-30% of people — which is why diazepam (Valium) was for many years the most frequently prescribed drug in North America. Alcohol & barbiturates have similar effects on the GABA receptor. In fact, potentiation of chloride influx into neurons is a major mechanism in the effect of ethanol on the brain. Some of the effects of benzodiazepines are probably due to GABA synapses on monoamine-producing neurons. GABA receptors can also be blocked, and the insecticide dieldrin is used for this purpose.
Prolonged use of benzodiazepines results in adaptation of the receptors to their use. Receptors may increase in number and/or sensitivity to GABA. (An increase or decrease in receptor number or sensitivity due to receptor alteration by drugs is known as upregulation or downregulation, respectively. A larger dose of benzodiazepine may be needed to produce the same result — a phenomenon known as tolerance. Withdrawal of the drug can result in GABA receptor hypoactivity producing symptoms worse than the ones that the patient originally sought treatment for. Such symptoms are called withdrawal. The phenomenon of receptor adaptation and drug dependence is seen with most drugs that act at synapses, including ones that are excitatory or potentiating as well as inhibitory or deactivating.
VIII. ACETYLCHOLINE
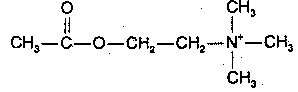
|
Acetylcholine was the first neurotransmitter discovered and is the major neurotransmitter in the peripheral nervous system (the only other peripheral neurotransmitter being norepinephrine). Acetylcholine is usually (but not always) an exitatory neurotransmitter — in contrast to the monoamine neurotransmitters, which are nearly always (with a few exceptions) inhibitory. Acetylcholine in the brain is produced from acetyl-CoA, resulting from glucose metabolism, and from choline, which is actively transported across the blood-brain barrier. Most dietary choline comes from phosphatidyl choline, the major phospholipid in the membranes of plants&animals (but not bacteria). The acetyl-CoA & choline are independently synthesized in the neuron cell body and independently transported along the axon to the synapse where they are conjugated into acetylcholine.
There are comparatively few acetylcholine receptors in the brain,
but outside the brain acetylcholine is the major neurotransmitter controlling
the muscles. Body muscles can be divided into the skeletal muscles system
(under voluntary control) and the smooth muscles of the autonomic nervous
system (controlling heart, stomach,
etc. — not
under voluntary control). The autonomic nervous system is further subdivided
into sympathetic and parasympathetic divisions. Direct innervation
of skeletal muscles is due to acetylcholine, as is the innervation of smooth
muscles of the parasympathetic nervous system. Direct innervation of the
sympathetic nervous system (except for sweat glands) is due to norepinephrine
(or both epinephrine & norepinephrine in the case of the adrenal medulla).
![[Sympathetic and Parasympathetic Nervous Systems]](./FigX12.gif)
|
The sympathetic nervous system innervates body organs in "fight or flight" situations, so the role of norepinephrine as the end-organ neurotransmitter should not be surprising. End-organ stimulation by acetylcholine in the parasympathetic nervous system is more "vegetative", eg, assisting digestion. Acetylcholine receptors are of two types: (1) a fast-acting ion-channel controlled receptor and (2) a slow-acting receptor that acts through a G-protein (Guanine nucleotide-binding protein) that stimulates second-messengers (often cyclic AMP) to indirectly open ion-channels. Direct ion-channel controlling receptors can respond in microseconds, whereas indirect second-messenger controlling receptors take milliseconds to produce a response. Only indirect, second-messenger controlling receptors have the capacity for plasticity. The two acetylcholine receptor classes are named for artificial toxins that selectively activate them. The fast-acting receptor is named nicotinic, because it is specifically activated by the toxin found in tobacco. The slow-acting receptor is named muscarinic, because the toxin muscarine (found in poisonous mushrooms) and acetylcholine will activate it, but nicotine will not.
Parasympathetic nerves are either cranial or sacral. 75% of all parasympathetic fibers arise from a single cranial nerve: the vagus nerve. These fibers travel to end-organs containing ganglia. It is the short postganglionic nerves from the ganglia to the smooth muscles in the end-organs which are muscarinic. The preganglionic fibers are nicotinic. Similarly, the preganglionic fibers of the sympathetic nervous system are nicotinic, although the sympathetic ganglia exist as distinct nodules closer to the spinal cord. The neuromuscular junction of skeletal muscles is also nicotinic.
Considering the rapidity with which skeletal muscles must often be able to respond to the volition to move, it is understandable that they are controlled by fast-acting nicotinic receptors. Unlike other neurotransmitters (which rely on re-uptake), acetylcholine activity in both muscarinic and nicotinic synapses is primarily stopped by an enzyme, ie, acetylcholinesterase. For nicotinic synapses, this means that a signal can be both rapidly initiated and rapidly terminated. The choline resulting from the hydrolysis of acetylcholine can be transported across the presynaptic membrane for resynthesis into acetylcholine.
Some snake venoms contain toxins that block nicotinic receptors,
thereby paralyzing their victims. Similarly, some South American Indians
used the nicotinic blocking agent curare (extracted from plants) as a
poison on their arrowheads.
Atropine, which blocks muscarinic receptors, is also a poison.
But atropine-like substances are of use for dilating the
eye through topical application, for examination of the retina.
![[cholinergic pathways in the brain]](./FigX13.gif)
Not only are there relatively few cholinergic neurons in the brain, but their distribution is "spotty" — in contrast to the monoamines which have distinct midbrain nuclei serving as the major sources of brain innervation. Most brain cholinergic receptors are muscarinic, which may make sense insofar as only second-messenger controlled receptors are capable of synaptic plasticity. The site of greatest acetylcholine synthesis in the brain is the interpeduncular nucleus (located near the substantia nigra in the midbrain). All of the interneurons in the striatum (caudate nucleus & putamen of the basal ganglia) and the nucleus accumbens are cholinergic. The septum provides cholinergic fibers to the septal-hippocampal tract. The primary cholinergic input to the cerebral cortex comes from the basal nucleus of Meynert, which is the most prominent structure in the substantia innominata (ventral to the anterior half of the globus pallidus, and adjacent to the hypothalamus). Meynert's nucleus also innervates the basolateral amygdala, the basal ganglia and the reticular nucleus of the thalamus. Cholinergic input from the basal nucleus to the cerebral cortex is active in both the waking state and in REM sleep, but is reduced in non-REM sleep.
Administration of centrally-acting muscarinic blocking agents results in memory loss for normal individuals. The fact that Alzheimer's Disease patients show severe reduction in Meynert's nucleus neurons has led to the suspicion that cholinergic loss could be an important factor in memory loss for these patients. Benefits from the anticholinesterase drugs have reinforced this belief. But Alzheimer's patients have neurofibrillary tangles, loss of NMDA receptors and reduced numbers of noradrenergic & serotonergic neurons as well. Some Alzheimer's patients show normal cholinergic cell numbers in Meynert's nucleus.
Centrally-acting anticholinesterases such as physostigmine (which can cross the blood-brain barrier — in contrast to neostigmine, which cannot) will worsen the tremors seen in Parkinson's Disease. Conversely, atropine-like substances (which are anti-muscarinic) reduce those tremors. These effects are clearly a consequence of the cholinergic interneurons in the striatum. Physostigmine & neostigmine are known as reversible anticholinesterases because they bind to acetylcholinesterase, but will eventually disengage. This makes them of therapeutic use for glaucoma & myasthenia gravis. Irreversible anticholinesterases which permanently bind-to and inactivate acetylcholinesterase, are only of use as insecticides. The only treatment for poisoning by these substances is atropinic drugs.
Stimulation of brain nicotinic receptors by nicotine from tobacco and arecoline from betal nuts (chewed by a quarter of the population of India) is a source of euphoric addiction for a large segment of the world's population. Research funded by the tobacco industry has shown that working memory may be improved by nicotinic stimulation of dopaminergic neurons in the substantia nigra & ventral tegmental area [BRAIN RESEARCH 657:165-170 (1994)] and that nicotine may be able to improve learning & memory in Alzheimer's patients showing a loss of nicotinic receptors in the neocortex & hippocampus [PHARMACOLOGY BIOCHEMISTRY AND BEHAVIOR 52:517-523 (1995)].
IX. DOPAMINE
The primary monoamine neurotransmitters are dopamine, norepinephrine and serotonin. Dopamine and norepinephrine are catecholeamines, whereas serotonin is an indolamine.
The amino acid tyrosine is not an essential amino acid because it
can be synthesized in the liver from phenylalanine by the enzyme
phenylalanine hydroxylase. But it cannot be synthesized in the
brain, and therefore must enter the brain by the large neutral amino
acid transporter, which also transports phenylalanine, tryptophan, methionine
and the branch-chained amino acids. These amino acids all compete for the
transporter, so a large quantity of one of the other amino acids in the
blood stream could greatly limit the amount of tyrosine entering the brain.
One case in which this occurs is when there is a liver deficiency of
phenylalanine hydroxylase. In that case, Phenylalanine reaches high
concentrations in the blood and monopolizes the large neutral amino acid
transporter, producing the mental retardation of phenylketonuria.
![[Dopamine synthesis]](./FigX14.gif)
|
Once in the brain, tyrosine can be converted to DihydrOxyPhenylAlanine (DOPA) by the tyrosine hydroxylase enzyme using oxygen, iron and TetraHydroBiopterin (THB) as co-factors. High concentrations of dopamine inhibit tyrosine hydroxylase activity through an influence on the THB co-factor. DOPA is converted to dopamine by Aromatic Amino Acid Decarboxylase (which is fairly nonspecific insofar as it will decarboxylate any aromatic amino acid) using PyridoxaL Phosphate (PLP) as a co-factor. This reaction is virtually instantaneous unless there is a Vitamin B6 deficiency.
Dopamine & epinephrine are primarily inhibitory neurotransmitters that
produce arousal. This may sound paradoxical, but the most likely explanation
for this effect is that the postsynaptic cells for catecholamines
themselves are inhibitory. There are 3-4 times more dopaminergic cells in
the CNS than adrenergic cells. Dopamine in the caudate nucleus facilitates posture,
whereas dopamine in the nucleus accumbens is associated with an animal's speed
(and pleasure).
![[dopamine receptors]](./FigX15.gif)
There are two primary dopamine receptor-types: D1 (stimulatory) and D2 (inhibitory), both of which act through G-proteins. D2 receptors often occur on the dopaminergic neurons, partially for the purpose of providing negative feedback. These so-called autoreceptors can inhibit both dopamine synthesis and release.
The binding of dopamine to D1-receptors stimulates the
activity of Adenylyl Cyclase (AC), which converts ATP to cyclic
AMP (cAMP), a second
messenger which binds to Protein Kinase A (PKA). PKA then
modulates the activity of various proteins by the addition of phosphate.
![[dopamine pathways]](./FigX16.gif)
There are 4 main dopaminergic tracts
in the brain: (1) the nigrostriatial
tract from the substantia nigra to the
striatum accounts for most of the brain's
dopamine (2) the tuberoinfundibular tract
from the arcuate nucleus of the hypothalamus
to the pituitary stalk, which has a controlling
effect on the release of the hormones
prolactin through tonic inhibition via
D2 receptors (3) the mesolimbic tract
from the ventral tegmental area to many
parts of the limbic system and (4) the
mesocortical tract from the ventral
tegmental area to the neocortex,
particularly the prefrontal area.
Dopamine cells project topographically to
the areas they innervate.
![[monoamine metabolism]](./FigX17.gif)
Both dopamine & norepinephrine are catabolized by a two-step process involving the enzymes MonoAmineOxidase (MAO) and Catechol-O-MethylTransferase (COMT). COMT is primarily active in the synapses, and uses &S-Adenosyl Methionine (SAM) as a methyl-group donor. MAO is primarily active in the pre-synaptic terminal against catecholamines that are not safely enclosed in storage vesicles. Normally, COMT only catabolizes about 10% of synaptic catecholamine, since catecholamine synaptic activity is primarily terminated by re-uptake into the pre-synaptic neuron terminal. MAO accounts for a much larger portion of catecholamine metabolism.
The darkly pigmented neurons in the pars compacta of the substantia nigra accounts for 80% of the dopamine in the brain. The dark pigment neuromelanin is a dopamine polymer that makes the substantia nigra appear black. Motor control in the striatum (caudate nucleus and putamen) is thought to involve a balance between inhibitory dopaminergic (D2) and excitatory cholinergic neurons.
A form of MAO, known as MAO-B, is the most common form of MAO in the
striatum. MAO-B is known to metabolize the neurotoxin MPTP to its active
form. When striatum dopamine is depleted to 20% the original level,
symptoms of Parkinson's Disease appear. Administration of DOPA is the
most common treatment. The monoamine oxidase inhibitor
deprenyl
will specifically inhibit MAO-B, thereby making deprenyl a useful adjunct to
DOPA therapy. The ergot derivative bromocriptine is a D2 agonist which
can alleviate the symptoms. (Deprenyl's reputed anti-aging capabilities may be related to MAO-B inhibition. Similarly,
carnosine, which has been
shown to reduce cellular senescence, inhibits MAO-B.)
![[Phenothiazine derivatives]](./FigX18.gif)
|
Schizophrenia is thought to be due to an overstimulation of D2 receptors in the mesolimbic and mesocortical systems. Evidence for the "excess dopamine" theory of schizophrenia comes largely from the fact that D2 antagonist drugs alleviate the symptoms, whereas substances which increase D2 stimulation, such as amphetamines, can induce psychotic symptoms (which are reversible with D2 antagonists). About 10% of Parkinsonian patients given DOPA treatment will develop psychotic symptoms resembling schizophrenia.
The major classes of antipsychotic drugs are the phenothiazines (eg, chlorpromazine), the butyrophenones (eg, haloperidol) and the thioxanthenes (eg, chlorprothixene). Butyrophenones are 100 times more potent against D2 receptors than against D1 receptors. The similarity in shape between a portion of the chlorpromazine molecule and dopamine indicates how chlorpromazine could bind to a dopamine receptor without triggering a response.
The mesolimbic & mesocortical dopaminergic systems are thought to play an important role in motivation, by attaching cognition of incentive significance to stimuli. In experiments on animals that are motivated to electrically self-stimulate themselves with electrodes implanted in their brains, dopamine is the mediating neurotransmitter for the locus ceruleus, lateral hypothalamus, ventral tegmental area and sulcal prefrontal cortex (but not the nucleus accumbens or substantia nigra). Fruit flies that are "socially stimulated" have three times the amount of dopamine in their brains than "socially deprived" fruit flies — an effect that correlates directly with more sleep (presumably consolidating learning) in the stimulated flies [SCIENCE; Indrani,G; 313:1775-1781 (2006)].
Cocaine particularly increases dopaminergic activity in the mesolimbic areas of the brain by inhibiting dopamine re-uptake in the ventral tegmental area and the nucleus accumbens. Amphetamine seems more generalized in its action, not only by inhibiting re-uptake, but by releasing dopamine from most brain regions. Both cocaine & amphetamine produce feelings of psychological energy & arousal, associated with diminished appetite & need for sleep. Both cocaine & amphetamine can lead to visual & tactile hallucinations as well as paranoid thinking, although the psychotic effects of amphetamine may also be mediated by increased serotonin release. Chronic amphetamine users seem to lose a capacity for normal pleasure — which has been correlated with neuron degeneration in the mesolimbic area.
Perception of time-intervals is believed to be mediated by spiny neurons located in the striatum of the basal ganglia. Timing begins with a burst of dopamine and ends with a recognized signal. Marijuana slows subjective time by lowering dopamine available, whereas cocaine and methamphetamine accelerates the sense of time by increasing dopamine availability. (Adrenaline and stress hormones can also "make seconds feel like hours".)
The natural brain amine phenylethylamine (PEA, found in chocolate) has been associated with the "love-excitement" of sexual attraction & emotional infatuation.
PEA concentrations are normally highest in the nucleus accumbens (a "reward center") followed by the frontal & cingulate cortices. Levels spike during orgasm and ovulation. PEA is very similar to amphetamine in chemical structure and may likewise act by causing dopamine release, but endorphin release may be a significant effect. PEA is preferentially oxidized by MonoAmine Oxidase-B (MAO-B), which may account for the anti-depressant effects of selegiline.
Other actions of dopamine include the induction of vomiting by stimulation of D2 cells in the chemoreceptor trigger zone, stimulation of growth hormone release by D2 receptors, and increased exploration & locomotion (thought to be connected to dopaminergic activity in the nucleus accumbens). Sexual behavior in the male is increased by dopamine agonists, whereas sexual behavior in the female is increased by dopamine antagonists.
X. NOREPINEPHRINE (NORADRENALIN)
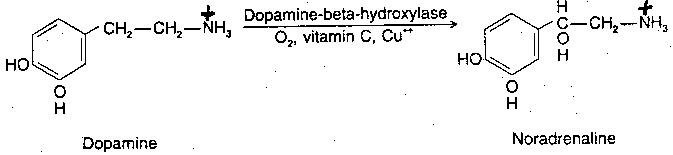
|
Norepinephrine (along with acetylcholine) is one of the two neurotransmitters in the peripheral nervous system. Norepinephrine is synthesized from dopamine by means of the enzyme Dopamine Beta-Hydroxylase (DBH), with oxygen, copper and Vitamin C as co-factors. Dopamine is synthesized in the cytoplasm, but norepinephrine is synthesized in the neurotransmitter storage vesicles. Cells that use norepinephrine for formation of epinephrine use SAMe (S-AdenylMethionine) as a methyl group donor. Levels of epinephrine in the CNS are only about 10% of the levels of norepinephrine.
The most prominent noradrenergic (ie, norepinephrine-containing)
nucleus is the locus ceruleus in the pons,
which account for over 40% of noradrenergic neurons in the rat brain. Most
of the other noradrenergic neurons are clustered in a region described as
the lateral tegmental area. The neocortex, hippocampus, and cerebellum
receive noradrenergic stimulation exclusively from the locus ceruleus. Most
of the dopaminergic innervation of the hypothalamus comes from the lateral
tegmental nuclei.
![[noradrenergic pathways in the brain]](./FigX20.gif)
Electrical stimulation of the locus ceruleus produces a state of heightened arousal. The noradrenergic system is most active in the awake state, and it seems to be important for focused attention, in contrast to the motor arousal of dopamine. Although the locus ceruleus has been identified as a pleasure center, it also seems to contribute to anxiety. Increased neuronal activity of the locus ceruleus is seen upon the occurrence of unexpected sensory events. Brain norepinephrine turnover is increased in conditions of stress. Benzodiazepines, the primary antianxiety drugs, decrease firing in the locus ceruleus, thus reducing distribution of noradrenalin to the forebrain and amygdala. This is part of the explanation for the use of benzodiazepines for inducing sleep.
Active projection of norepinephrine from the locus coeruleus of the reticular activating system to the forebrain is a key feature of awakeness-arousal as distinguished from sleep. Norepineprhine projection to the basal nucleus of the forebrain is low in sleep — virtually absent in REM (Rapid Eye-Movement) sleep. The basal nucleus when stimulated by norepinephrine from the locus coeruleus sends neuromodulating acetylcholine to the cerebral cortex, thereby promoting alertness.
The beta-adrenergic blocking drug propranolol has also been used to treat anxiety. By blocking the adrenergic inputs to the amygdala, beta-blockers inhibit the formation of traumatic memories. Cortisol stimulation of the locus coeruleus due to chronic stress exacerbates norepinephrine stimulation of the amygdala. Healthy humans fed fish oil rich in the omega-3 fatty acids EPA and DHA show reduced plasma levels of the stress-associated hormone norepinephrine [NUTRITION; Hamazaki,K; 21(6):705-710 (2005)].
Beta-noradrenergic receptors also apparently inhibit feeding, whereas alpha-receptors seem to stimulate feeding.
Although MAO inhibitors reduce metabolism of all catecholamines, it
is believed that the anti-depressant effect is more related to norepinephrine
than to dopamine. Most MAO in the brain is of type-B, but drugs selective
for inhibiting MAO-A have proven to be better anti-depressants. MAO-A
preferentially metabolizes norepinephrine & serotonin. MAO-A inhibiting
drugs given for depression have critically elevated blood pressure in patients
eating tyramine-containing foods (such as cheese) due to the failure to
metabolize tyramine (which can act as a pressor agent). These drugs
(eg, phenelzine & pargyline) inactivate
MAO by forming irreversible covalent bonds. More modern MAO inhibitors are
safer because they form reversible bonds. MAO-B inhibitors like deprenyl are
also less likely to cause the "cheese effect". (Alcohol also selectively
inhibits MAO-B.)
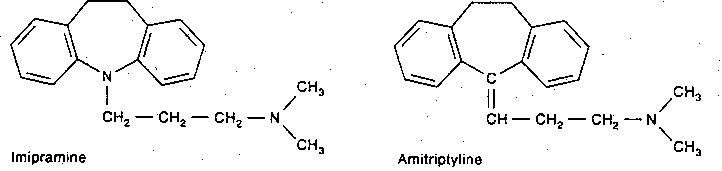
|
Tricyclic anti-depressants derive their name from their 3-ring structure. Desipramine only inhibits norepinephrine re-uptake, with little effect on dopamine. Imipramine & amitriptyline are inhibitors of norepinephrine and serotonin re-uptake by the presynaptic terminals, but are more potent for serotonin. Cocaine is also a potent inhibitor of catecholamine re-uptake, but it does not act as an anti-depressant. Weight gain due to increased appetite is a frequent side effect of tricyclic anti-depressants, particularly of amitrip- tyline. By contrast, both cocaine & amphetamine reduce appetite.
Both MAO inhibitors and tricyclic anti-depressants have immediate effects on brain monoamines, but clinically anti-depressants require several weeks of administration before they produce a therapeutic effect. It is therefore believed that it is not the immediate effects on neurotransmitters that is producing the antidepression, but the long-term effects on modification of receptors.
Excessive cortisol secretion is seen in 40-60% of depressed patients, associated with diminished noradrenergic inhibition of corticotropin-releasing hormone secretion in the hypothalamus. Corticotropin-releasing hormone induces anxiety in experimental animals.
XI. SEROTONIN (5-HYDROXYTRYPTAMINE, 5-HT)
![[indole molecule]](./FigX22.gif)
Serotonin was isolated from the blood serum
as a substance causing powerful smooth muscle contraction.
Only later was it demonstrated to be tryptamine with a
hydroxyl group at the 5-position. Only 1-2% of the
serotonin in the body is in the brain, insofar as serotonin
is widely distributed in platelets, mast cells, etc. But
there is no equilibration between body serotonin and brain
serotonin — the serotonin in the brain is independently
synthesized from tryptophan transported across the blood-brain barrier.
![[Serotonin synthesis]](./FigX23.gif)
|
Serotonin synthesis is a 2-step process, the first step of which requires the enzyme tryptophan hydroxylase with oxygen, iron and THB as co-factors. Neither the enzyme nor the co-factors are rate-limiting for either step of these reactions — virtually all brain tryptophan is converted to serotonin. Serotonin concentration in the brain is far more sensitive to the effects of diet than any other monoamine neurotransmitter — and can be increased up to 10-fold by dietary supplementation in laboratory animals.
Consumption of a meal that is high in carbohydrate, branch-chained amino
acids and tryptophan has a particularly dramatic effect because both glucose
from carbohydrate and branch-chained amino acids (especially leucine) increase
insulin secretion. Insulin facilitates the transport of the branch-chained
amino acids into muscle cells, thereby reducing the competition tryptophan
faces for the large neutral amino acid transporter that takes it across the
blood-brain barrier. The resultant drowsiness induced by serotonin is a
common effect of a large carbohydrate meal.
![[Melatonin]](./FigX24.gif)
|
The richest concentration of serotonin in the body can be found in the pineal body, even though this gland does not use serotonin as a neurotransmitter. Instead, serotonin is primarily methylated in the synthesis of melatonin. Melatonin derives its name from the fact that it can darken the skin of amphibians ("melas" is Greek for "black") — although it has also been reported to induce pigment lightening in cells. Melatonin is synthesized from serotonin in a 2-step process that takes an acetyl group from acetyl-CoA and a methyl group from by SAMe (S−Adenosyl Methionine).
Melatonin
is of particular importance for regulating diurnal (circadian)
& seasonal behavior & physiology in mammals. The pineal body has been called
a "third eye" because its activity is influenced by light. In mammals,
noradrenergic neurons near the optic nerve are inhibited by light. In
darkness, norepinephrine stimulation of pineal cells causes the release
of cyclic AMP second-messenger, which activates (phosphorylates) the
N-acetyl transferase enzyme which catalyzes acetylation of serotonin.
In many specied (including humans) melatonin is an inhibitor of sexual activity in both sexes. Decreased
melatonin in the Spring leads to rutting — and the birth of offspring in
the warmer seasons. Melatonin also stimulates production of brown adipose
tissue, a special form of fat which (when burned) only produces heat, not
ATP. This is especially important for hibernating animals.
![[serotonin pathways in the brain]](./FigX25.gif)
Serotonin neurotransmitter neurons are located in the raphe nuclei. The caudal (closer to the "tail") nucleus projects largely to the medulla and spinal cord for the regulation of pain perception. The rostral (closer to the "beak") nucleus projects extensively to the limbic structures and the cerebral cortex. In the limbic system, especially, the projections are co-localized with norepinephrine receptors — and the two transmitters seem to work in conjunction in the regulation of arousal.
The SupraChiasmatic Nucleus (SCN) of the hypothalamus regulates the mammalian circadian clock ("day-night cycles"), partially in response to light. Melatonin release is inhibited as a result of the response of the SCN to light. The SCN is richly innervated by serotonergic input from the dorsal raphe nucleus. Serotonin inhibits the responsiveness of the SCN (and thus the circadian rhythm) to light [ANNALS OF MEDICINE 31:12-33 (1999)]. Sleep deprivation increases serotonin release in the SCN [BRAIN RESEARCH 909:81-91 (2001)]. With aging there is a decline in both serotonin transporters [LIFE SCIENCES; Yamamoto,M; 71(7):751-757 (2002)] and serotonin receptors [NEUROPSYCHOPHARMACOLOGY; Meltzer,MD; 71(7):751-757 (2002)]. Depletion of serotonin is believed to be related to the disruption of the circadian rhythm associated with senescence [AMERICAN JOURNAL OF PHYSIOLOGY 272(2 Pt 2):R509-R513 (1997)].
As little as one or two grams of L−tryptophan is effective in decreasing sleep latency time [PSYCHOPHARMACOLOGY; Schneider-Helmert,D; 89(1):1-7 (1986) and PHARMACOPSYCHIATRY; Demisch,K; 20(6):242-244 (1987)]. L−tryptophan either improves or has not effect on other sleep parameters, with the exception of a suppressive effect on REM sleep [EUROPEAN NEUROLOGY; Korner,E; 25(Suppl 2):75-81 (1986)].
Almost all antidepressant treatments (tricyclic antidepressants, monoamine oxidase inhibitors, lithium, electoconvulsive therapy, serotonin reuptake inhibitors, etc.) augment serotonin neurotransmission [MOLECULAR PSYCHIATRY; Bauer,M; 7(2):140-156 (2002)]. Low levels of serotonin are associated with high levels of pain sensitivity, locomotion, aggression, and sexual activity [BIOLOGICAL PSYCHIATRY; Lucki,I; 44(3):151-162 (1998)]. Low levels of serotonin are also associated with depression, panic disorders, and Obsessive-Compulsive Disorder (OCD). Patients with evidence of low serotonin levels have attempted suicide by very dramatic means, such as cutting the throat. This may explain some of the therapeutic effects of fluoxetine (Prozac), which selectively prevents the re-uptake of serotonin, thereby increasing synaptic serotonin concentrations (elevated serotonin). Fluoxetine is also distinctive because it has a half-life of about four days. Fluoxetine has been used therapeutically for panic, obsessive-compulsive and eating disorders (such as bulimia). Unlike the tricyclic anti-depressants, which often stimulate appetite, fluoxetine more often reduces appetite. Fluoxetine may even enhance learning [PHARMACOLOGY BIOCHEMISTRY AND BEHAVIOR 52:341-346 (1995)]. Depression patients treated with tryptophan as well as fluoxetine show less sleep disturbance at the outset of treatment than patients treated with fluoxetine alone [JOURNAL OF PSYCHIATRY & NEUROSCIENCE; Levitan,RD; 25(4):337-346 (2000)].
Rats subjected to learned helplessness conditioning (electric shocks) became desensitized to serotonin, but exercise (running) reduces this effect [JOURNAL OF NEUROSCIENCE; Greenwood,BN; 23(7):2889-2898 (2003)]. Depression of hippocampal neurogenesis by learned helplessness conditioning in rats can be reversed by fluoxetine [NEUROPSYCHOPHARMACOLOGY; Malberg,JE; 28(9):1562-1571 (2003)].
The natural carbohydrate inositol has been used to elevate serotonin without some of the side effects of fluvoxamine (Luvox, which is similar to Prozac). A dose of 18 grams/day inositol has been shown to be effective against Obsessive-Compulsive Disorder (OCD) [THE AMERICAN JOURNAL OF PSYCHIATRY; Fux,M; 153(9):1219-1221 (1996)]. The same dose of myo-inositol has been shown to be as effective as flavoxamine against panic disorders [JOURNAL OF CLINICAL PSYCHOPHARMACOLOGY; Palatnik,A; 21(3):335-339 (2001)]. Cantaloupe, oranges, and grapefruit are the foods containing the highest inositol concentrations, but to get 18 grams/day inositol would require eating 13 cantaloupes, 45 grapefruits, or 58 oranges [AMERICAN JOURNAL OF CLINICAL NUTRITION; Clements,RS; 33(9):1954-1967 (1980)].
The tricyclic antidepressants typically inhibit both norepinephrine & serotonin re-uptake by pre-synaptic terminals. The effectiveness of these neurotransmitters against depression seems to be due to both decreased functional activity of beta-postsynaptic norepinephrine receptors and increased activity of type−2 serotonin receptors in the limbic regions of the brain. But experimental depletion of monoamines only leads to depression in subjects having a family history of this disorder [MOLECULAR PSYCHIATRY; Ruhe,HG; 12(4):331-359 (2007)].
Monkeys with high levels of testosterone & low levels of serotonin are both aggressive & lacking in restraints on impulsive/violent behavior. Arsonists who commit their crime for mercenary reasons show normal levels of serotonin, but those who commit the crime impulsively have low serotonin. Lead interferes with serotonin synapse formation. Monkeys experimentally exposed to lead became so dangerously aggressive that the study was halted early [CHEMICAL & ENGINEERING NEWS 81(22):33-37 (2003)].
Reserpine prevents the transport of all the monoamines (and acetylcholine) into storage vesicles in the presynaptic membrane — leaving them vulnerable to destruction by monoamine oxidase. Reserpine (as extracts from the Rauwolfia plant) was used for centuries in India to treat "hysteria". Reserpine has been used as a potent tranquilizer, but it can produce serious depression that may lead to suicide attempts.
LySergic acid Diethylamine (LSD) acts most strongly on the type-2 serotonin receptors, but it also has some effect on norepinephrine receptors. Serotonin seems to play a role in dreaming. During both dreaming and LSD intoxication, electrical activity in the visual cortex arises from the brain stem rather than from the eyes. LSD not only induces visual hallucinations, but it heightens sensory awareness while diminishing control of sensory input. The reduced ability to distinguish between sensory impressions can lead to feelings of being "in union with the universe". Artificial stimulation of the raphe simulates the actions of LSD, decreasing habituation to repetitive stimuli. Low doses of LSD & amphetamine, however, have been shown to enhance a form of associative learning.
High-estrogen contraceptives may have contributed to depression by lowering serotonin levels in the brain. Low levels of growth hormone in depressed patients may be due either to low levels of norepinephrine, serotonin, or both.
XII. PEPTIDES
Peptides are the most common neurotransmitters in the hypothalamus. Their complex structure can allow for high receptor specificity. They are all synthesized on ribosomes and are all inactivated by hydrolysis at the synapse (rather than by re-uptake). Peptides are far more potent than other neurotransmitters, requiring only very small amounts to produce a profound effect. Even very minute amounts of somatostatin can inhibit growth hormone release.
Opioid peptides include the endorphins, enkephalins and dynorphins. Enkephalins are frequently found in presynaptic (axo-axonic) synapses. Opiates and enkephalins (or endorphins) inhibit the firing of locus ceruleus neurons. The highest concentration of opioid receptors are found in the sensory, limbic and hypothalamic regions of the brain — and are particularly high in the amygdala & periaqueductal grey area. Opioids tend to be released as slower-acting co-transmitters which modulate the action of the associated neurotransmitter (such as glutamate) which is being released from the same synapse. Although opioids are generally inhibitory, they have an excitatory effect on hippocampal pyrimidal neurons mediated by inhibition of GABA release.
Cholecystokinin (CCK) seems to function in the production of satiety. Injection of small quantities of this peptide into the ventricles or the paraventricular nucleus can inhibit feeding. CCK is associated with dopamine synapses in some limbic areas, and appears to modulate dopamine release. Such peptide synergy with other transmitters is common. For example, GABA is often associated with somatostatin and serotonin with Substance P.
Low doses of the peptide vasopressin have been shown to enhance learning in laboratory animals. However, humans with vasopressin deficiency show no signs of memory impairment. Because vasopressin is potent in increasing blood pressure, its use by humans should be approached with caution. Safer analogues may yet be found.
XIII. RELEVANCE TO CRYONICS
The profound effects of neurotransmitters on the human psyche seems to portend substantial implications for personal identity. But their obvious emphemeral nature makes it seem unlikely that they can be described as being part of "the anatomical basis of mind". Water is also an important component of the human brain, but water is very uniform and replaceable. Preservation of the means of producing, storing, releasing and re-uptaking neurotransmitters (ie, the receptors and synaptic connections) seems more important than preserving the neurotransmitters themselves. The fact that anti-depressants & anti-psychotics take weeks to become effective may offer some clue in this regard.
In a sense, however, many of the "structural" features of neurons are no less ephemeral than neurotransmitters. They can be produced or replaced by enzymes coded-for by DNA in much the same way as neurotransmitters. This would seem to point to neuron gene expression as the key locus for identity, and the most crucial site for preservation. But gene expression is determined by the mileu of the neuron — a circular homeostasis that seemingly does not allow for finding "ultimate causes". Moreover, considering the demonstrably critical role played by astrocytes in GABA re-uptake at synapses, one might wonder whether glial cells are also an essential component of identity.
(For more on the subject of the locus of consciousness in the brain and the relevance to preservation of consciousness see my essay Neurophysiology and Mental Function.)
![[GO TO BEN BEST'S HOME PAGE]](../../homeback.gif) HOME PAGE
HOME PAGE