
According to the "immune system theory of aging", many aging effects are due to the declining ability of the immune system to differentiate "foreign" from "self" proteins. Not only does the immune system become less capable of resisting infection & cancer, but declining cell function could be due to attacks by the immune system against native tissues. Arthritis, psoriasis and other autoimmune diseases increase with age. There is evidence that histocompatability genes, genes affecting DNA repair and genes for SOD production — all of which affect longevity — are located close together on human chromosome 6.
Leukocytes (white blood cells), which form the basis of the immune system (along with complement proteins), are roughly 65% granulocytes (mostly neutrophils), 5% monocytes (which can become macrophages) and 30% lymphocytes. Lymphocytes can be subclassified as B−lymphocytes (B−cells) or T−lymphocytes (T−cells) based on whether they mature in Bone marrow or the Thymus gland (all lymphocytes originate in bone marrow). Antigens are molecular portions of pathogens that act as identifiers. B−cells generate antibodies ("humoral immunity") against antigens, whereas T−cells directly bind to antigens ("cellular immunity").
The thymus gland of the immune system reaches its greatest weight during puberty, and shrinks thereafter, with lymphoid tissue being replaced by fat. The shrinking of the thymus gland proceeds far more rapidly than the progress of aging — at age 50 the thymus of humans is typically only 5−10% of its original mass. Nonetheless, T−cells remain fairly constant over most of adult life due to peripheral proliferation (although proliferation declines in the elderly).
Because the thymus is the organ in which T−cells "matures", once maturation occurs most of the work of the thymus is done. In the maturing T−lymphocyte system, the thymus creates a broad diversity of T−cells, each of which is programmed to recognize and combat a different antigen. T−cells which would combat self-substances are eliminated by apoptosis.
The immune system uses proliferation & apoptosis to create & refine T−cells. The immune system uses clonal expansion (rapid multiplication of lymphocytes of a single "clone" against a single antigen) & apoptosis to control the numbers of T−cells available to fight specific antigen threats. Injecting the protein Apo−1 into a cell will trigger apoptosis. But the protein Bcl−2 can rescue a cell from apoptosis. With aging, mature T−cells increasingly manifest apoptosis for reasons that seem to be unrelated to decreased Bcl−2 expression or oxidative stress [MECHANISMS OF AGING AND DEVELOPMENT; Phelouzat,M; 88:25-38 (1996)].
T−lymphocytes that have not encountered an antigen since creation are called naive T−cells, whereas T−lymphocytes that have been clonally expanded to fight an invading antigen are called memory T−cells. T−cells of the elderly have a much higher ratio of memory T−cells to naive T−cells than younger people. Old memory T−cells have less CD28 surface protein than young memory T−cells and are thus less able to divide when presented with antigen [JOURNAL OF IMMUNOLOGY; Engwerda,CR; 152:3740-3747 (1994)]. CD28 ligation is required for production of IL−2 cytokines. The elderly memory T−cells have short telomeres and are thought to accumulate because of increasingly defective apoptosis [IMMUNOLOGIC RESEARCH 21(1):31-38 (2000)].
Two predominant forms of T−cells are cytotoxic T−cells (with CD8 surface receptors) and helper T−cells (with CD4 surface receptors). The cytotoxic T−cells attack bacteria or cancerous cells by punching holes in the cells and injecting them with toxic proteins. The helper T−cells secrete growth factors (cytokines) that foster the clonal expansion of other T−cells and/or of antibody-producing cells (the B−lymphocytes). Helper T−cells are more numerous in youth & maturity, but in the elderly the ratio of CD8 to CD4 cells increases. CD8 T−cells become more resistant to apoptosis with aging, whereas CD4 cells become more susceptible to apoptosis.
Cytomegalovirus prevalence significantly increases in the elderly, and may be responsible for much of the skewed CD8:CD4 ratio of advanced age [JOURNAL OF IMMUNOLOGY; Hadrup,SR; 176(4):2645 (2006)]. Cytomegalovirus infection increases with age in humans [CLINICAL INFECTIOUS DISEASES; Staras,SAS; 43(9):1143-1151 (2006)], primarily infects antigen-presenting cells & can increase inflammatory cytokines [REVIEWS IN MEDICAL VIROLOGY; Varani,S; 19(3):131-145 (2009)], and is associated with immunosenescence [CURRENT OPINION IN IMMUNOLOGY; Derhovanessian,E; 21(4):440-445 (2009)]. Naive CD4 cells decline rapidly after age 65 [JOURNAL OF IMMUNOLOGY; Naylor,K; 174(11):7446-7452 (2005)].
There are two types of helper T−cells, designated TH1 (type 1) and TH2 (type 2). The TH1 cells promote growth of T−lymphocytes with the cytokine InterLeukin−2 (IL−2), whereas the TH2 cells promote growth of B−lympocytes with the cytokine InterLeukin−4 (IL−4). TH1 cells are more prominent in autoimmune infections, whereas TH2 cells are more prominent in viral infections. In youth & maturity the TH1 cells predominate, but in the elderly the TH2 cells predominate [MECHANISMS OF AGING AND DEVELOPMENT 94:1-5 (1997)]. Moreover, aging is accompanied by a significant loss of IL−2 as well as of IL−2 receptors — a phenomenon thought to be responsible for the significant decline of proliferation (clonal expansion) in response to antigens seen with aging [SCIENCE 273:70-74 (1996)]. The decline of T−cell activation due to reduced IL−2 production is at least partially due to oxidation-damaged proteasomes being less capable of inducing the gene transcription factor NFκB [CELLULAR IMMUNOLOGY 192:167-174 (1999)].
Proliferation of T−cells in response to antigenic or mitogenic (cell-division stimulating) signals also declines with aging — apparently due to to decline in activity of the Mitogen Activating Protein Kinase (MAPK) cascade which causes cell surface signals to alter gene expression. CRAN (Caloric Restriction with Adequate Nutrition) significantly reduces the decline of MAPK activity associated with aging [PROCEEDINGS OF THE SOCIETY FOR EXPERIMENTAL BIOLOGY AND MEDICINE 223:163-169 (2000)]. But selenium supplementation has been shown to restore lymphocyte proliferation in aged mice to that of normal young adults [PROCEEDINGS OF THE SOCIETY FOR EXPERIMENTAL BIOLOGY AND MEDICINE; Roy,M; 209(4):369-375 (1995)].
The combination of low T−cell proliferation and low CD4/CD8 ratio was highly predictive of low 2−year survival in a study of people in the 86−92 age range [JOURNALS OF GERONTOLOGY 50A(6):B378-B382 (1995)]. Melatonin elevates the CD4/CD8 ratio [IMMUNITY & AGEING; Srinivasan,V; 2:17 (2005)]. Immune function is very important for the elderly because infection causes an increasing percentage of deaths for those over 80 years of age [JOURNALS OF GERONTOLOGY 52A(1):B67-B77 (1997)] and AMERICAN JOURNAL OF MEDICINE; 114:365-369 (2003)].
Regulatory T−cells that suppress T−cell-mediated autoimmune diseases in humans decline with age [JOURNAL OF NEUROSCIENCE RESEARCH; Tsaknaridis,L; 74(2):296-308 (2003)].
Natural Killer (NK) cells differ from cytotoxic T−cells by the ability to lyze pathogenic cells without the need of antigens. NK cells decline in activity with age, but this decline is compensated for by an increase in NK cell numbers. In centenarians, however, no decline in NK activity has been seen — nor was there a decline in youthful CD8/CD4 cells [IMMUNOLOGY TODAY; Franceschi,C; 16(1):12-16 (1995)].
B−cells from older animals produce less antibody and express less of the surface CD40 protein which causes B−cell activation and differentiation. The decline in T−cell activity with age is responsible for most of the decline in B−cell numbers and activity.
Macrophages are immune-system cells that "eat" foreign particles (including bacteria) and digest the particles in lysosomes. Monocytes are the small blood stream cells that swell to become macrophages after migrating into tissues. Monocytes from elderly humans have a greatly reduced capacity to produce the cytokine InterLeukin−1 (IL−1) and the toxic free radicals that macrophages use to kill foreign or cancerous cells [THE JOURNAL OF IMMUNOLOGY 154:832-843 (1995)]. Nonetheless, the superoxide, hydrogen peroxide, hydroxyl ions & nitric oxide produced by neutrophils & macrophages to kill bacteria can attack native tissues in age-associated chronic inflammation. The reactive products of nitric oxide and oxygen species inhibit PARP-mediated DNA repair [FREE RADICAL BIOLOGY & MEDICINE 35(11):1431-1438 (2003)].
Some of the decline in immune function in the elderly may be due to protein cross-linking in tissues & blood vessels reducing immune-cell mobility and access to infected areas. Poor nutrition in the elderly is also a factor. Supplements consisting of recommended dietary allowances of nutrients (plus extra Vitamin E & beta-carotene) significantly improved the immune status of elderly subjects [THE LANCET 340:1124-1127 (1992)]. Supplementation with the steroid hormone DeHydroEpiAndrosterone (DHEA, a hormone that dramatically declines with age) increased IL−2 & Interferon-gamma activity in mice [THE JOURNAL OF INFECTIOUS DISEASES 167:830-840 (1993)].
Vulnerability to death by influenza & pneumonia increases rapidly with age in the United States. A person aged 50−64 is nearly ten times more likely to die from an influenza-associated death as a person in the 5−49 age group. And a person over 65 is over ten times more likely to die from and influenza-associated death as a person in the 50−64 age group. A person over 85 is about 16 times more likely to die an influenza-associated death as a person in the 65−69 age group [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Thompson,WW; 289(2):179-186 (2003)]. Vaccination of the elderly reduces influenza-associated death by 50% [ARCHIVES OF INTERNAL MEDICINE; Hak,E; 165(3):274-280 (2005)].
With aging the body contains increasing quantities of proinflammatory cytokines such as TNF−α, IL−1 and IL−6, which is positively associated with cardiovascular disease mortality [IMMUNOLOGY AND ALLERGY CLINICS OF NORTH AMERICA; Bruunsgaard,H; 23(1):15-39 (2003)]. The increase in memory cells results in an increase in the cytokines IL−4 & IL−10 that are produced by the memory cells. Lifetime exposure to infectious disease reduces lifespan by accelerated immunosenescence [FEBS LETTERS; Martinis,MD; 579:2035-2039 (2005)] and chronic inflammation [SCIENCE; Finch,CE; 305:1736-1739 (2004)]. Chronic inflammation is implicated in atherosclerosis, arthritis, Alzheimer's Disease, cancer, the metabolic syndrome (type 2 diabetes) and numerous other afflictions affecting the elderly. Inflammation is probably not the major cause of the damage & degeneration of aging, but it contributes to the damage. Free radicals and oxidized glycation products (AGEs) are contributers to chronic inflammation.
|
|
Aging is associated with increasing activity of the pro-inflammatory transcription factor NF-κB (NF−κB). NF−κB is normally bound to IκB protein in the cytoplasm, but is released to enter the nucleus when infection, oxidative stress or pro-inflammatory cytokines cause ubiquitination and subsequent protease degradation of IκB. NF−κB increases transcription of genes coding for TNF−α and IL−1, which can result in a positive feedback loop. The ability of free radicals (ROS, Reactive Oxygen Species) to cause NF−κB release and the production of ROS by inflammation also results in a positive feedback loop. NF−κB and TNF−α are central to the aging-associated increase in chronic inflammation. Although glucocorticoids are increased in aging & CRAN and can inhibit NF−κB, stimulation of NF−κB by stressors predominates. Not only does NF−κB release increase with age, but aging results in NF−κB binding more strongly to DNA [BIOCHEMICAL JOURNAL; Helenius,M; 318(Pt 2):603-608 (1996)]. Age-associated increases in ceramide results in increased NF−κB activation [THE JOURNAL OF IMMUNOLOGY; Wu,D; 179(7):4829-4839 (2007)].
NF−κB induced chronic inflammation in combination with its ability to suppress apoptosis (inhibiting the elimination of cancer cells) often leads to cancer [NATURE IMMUNOLOGY; Karin,M; 3(3):221-227 (2002)]. Cancer can also be initiated by NF−κB induction of inducible Nitric Oxide Synthetase (iNOS), leading to DNA damage, and the inhibition of apoptosis by NF−κB again favors cancer [NATURE REVIEWS, IMMUNOLOGY; Karin,M; 5:749-759 (2005)].
|
Aside from the induction of TNF−α (Tumor Necrosis Factor-alpha) by NF−κB, TNF−α is produced by visceral fat. Obese people can produce twice as much TNF−α as lean people produce. White adipose tissue attracts macrophages, which produces the inflammatory agents (like TNF−α) associated with obesity-induced insulin resistance [JOURNAL OF CLINICAL INVESTIGATION; Xu,H; 112(12):1821-1830 (2003)]. Surgical removal of visceral fat (but not subcutaneous fat) extends the mean and maximum lifespan of rats [ BIOCHEMICA ET BIOPHYSICA ACTA; Huffman,DM; 1790(10):1117-1123 (2009)]. TNF−α can induce apoptosis, but only if protein synthesis is inhibited [SCIENCE; Beg,AA; 274:782-784 (1996)].
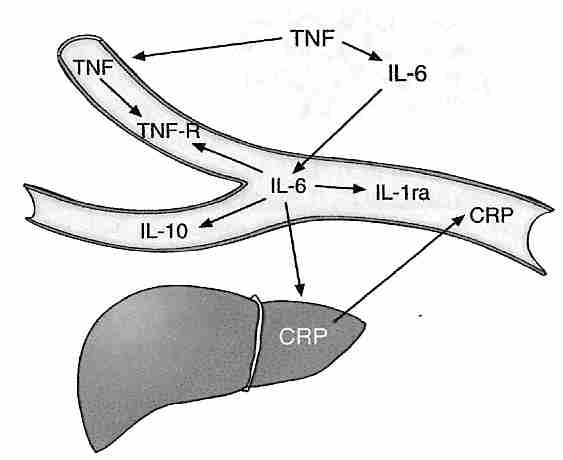
TNF−α upregulates NF−κB and IL−6 (InterLeukin−6). IL−6 upregulates pro-inflammatory cytokine IL−1 and induces the liver to produce the inflammatory protein CRP (C−Reactive Protein). Proinflammatory cytokines have been shown to induce cellular senescence [FREE RADICAL RESEARCH; Sasaki,M; 42(7):625-632 (2008) and CELL; Kuilman,T; 133(6):1019-1031 (2008)]. But IL−6 but also induces production of the anti-inflammatory cytokine IL−10 while inhibiting TNF−α. IL−10 (which inhibits TNF−α production) is produced in larger quantities when exogenous S-adenosylmethionine is administered [AMERICAN JOURNAL OF PHYSIOLOGY; Song,Z; 284(6):G949-G955 (2003)]. CRP is an important risk factor for myocardial infarction (heart attack). A four-year study of women showed those in the highest quarter of blood CRP had 15.7 times greater risk of developing type 2 diabetes as those in the lowest quarter [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Pradham,AD; 286(3):327-334 (2001)]. In a study of men, those in the highest quarter of blood CRP had 3 times the risk of developing dementia as those in the lowest quarter [ANNALS OF NEUROLOGY; Schmidt,R; 52(2):168-174 (2002)].
Increasing plasma levels of pro-inflammatory cytokines with aging can induce a stress response that is responsible for the increased plasma levels of cortisol associated with aging [INFLAMMATION RESEARCH; Sergio,G; 57(12):558-563 (2008)]. Although elevated cortisol is generally anti-inflammatory in the periphery, cortisol can be pro-inflammatory in the hippocampus and cerebral cortex [BRAIN, BEHAVIOR, AND IMMUNITY; Sorrells,SF; 21(3):259-292 (2007)].
Exercise can be very anti-inflammatory by increasing muscle-derived IL−6 production (which is independent of TNF−α) and reducing CRP [JOURNAL OF APPLIED PHYSIOLOGY; Peterson,AWW; 98(4):1154-1162 (2005)]. Adequate sleep can reduce TNF−α and IL−6 secretion (both of which induce sleepiness & fatigue). Reduction of IL−6 production by the administration of sex steroids has been suggested as a means of reducing problems with sleepiness & fatigue in the elderly [THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM; Vgontias,AN; 88(5):2087-2095 (2003)]. DHEA can also reduce IL−6 production [THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM; Straub,RH;83(6):2012-2017 (1998)].
CycloOXygenase (COX) enzyme activity increases with age, thereby increasing the production of prostaglandins that inhibit T−cell proliferation. Increased levels of hydrogen peroxide probably are responsible for the age-related increase in COX activity, indicated by the fact that Vitamin E attenuates COX activity and restores T−cell proliferation [AMERICIAN JOURNAL OF PHYSIOLOGY; Wu,D; 275(3 Pt 1):C661-C668 (1998)].
Advanced Glycation End-products (AGES) not only originate from metabolism, but can be ingested in diet or tobacco smoke and contribute significantly to inflammation. AGEs can activate NF−κB [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Vlassara,H; 99(24):15596-15601 (2002)]. High blood insulin potentiates NF−κB in a dose-dependent manner [CIRCULATION RESEARCH; Golovchenko,I;87(9):746-752 (2000)]. Increased NF−κB activity by AGEs is often mediated by a Receptor for AGE (RAGE), which can also be activated by TNF−α [JOURNAL OF BIOLOGICAL CHEMISTRY; Tanaka,N; 275(33):25781-25790 (2000)]. NF−κB activated by oxidative stress or AGEs upregulates the expression of RAGE (more AGE receptors), creating a positive feedback loop that worsens chronic inflammation [CIRCULATION RESEARCH; Schmidt,AM; 84(5):489-497 (1999)].
Even though age-associated chronic diseases are important components of segmental progerias , they are often dissociated from aging per se because those people who achieve maximum lifespan typically do not die of these diseases. The role of controllable risk factors like obesity, exercise, AGE ingestion and Vitamin E supplementation would also tend to dissociate inflammation and chronic disease from a central role in the essential aging process. Nonetheless, these chronics diseases are aging-associated and it is likely that inflammation plays some role in the degenerative & damaging processes known as aging.
Many chemicals accumulate in the cells with age, including toxic & inert substances from the exterior and similar substances arising as byproducts of cellular metabolism [notably Advanced Glycation End-products (AGEs) and lipid peroxidation debris]. Fat-soluble substances (such as DDT & PCBs) are particularly slow to be eliminated. Iron tends to accumulate in cell nuclei with aging, as does aluminum. Aluminum transforms metabolically active DNA into an inert state. Lead also accumulates in cells, and is neurotoxic. Cytochrome P−450 detoxification enzymes of the liver (which have maximal light absorption at 450 nanometer wavelength) decline with age. In the 1976 to 1980 period, the 15% of US population with the highest blood levels of lead had 49% higher cardiovascular mortality and 68% higher cancer mortality [ARCHIVES OF INTERNAL MEDICINE; Lustberg,M; 162(21):2443-2449 (2002)].
Non-dividing cells (muscle cells, heart muscle cells and neurons) are not susceptible to the Hayflick Limit. Nor is double-chromosome damage of as great concern in non-dividing cells as it is for dividing cells. But for non-dividing cells that cannot be replaced — heart muscle cells and neurons — the accumulation of cellular garbage may be a very significant factor in cellular aging. Species survival may be thus dependent on the creation of new organisms once the old ones have accumulated too much chemical garbage to be functional.
Of particular note is lipofuscin (age pigment), which can accumulate in large quantities in non-dividing cells. Lipofuscin is regarded as a product of lysosomes — organelles containing hydrolytic enzymes to degrade proteins, lipids and damaged organelles. As production of lysosome enzymes decline with age — and as lysosomes engulf increasingly cross-linked proteins & lipids that are resistant to enzyme degradation — dysfunctional lysosomes (bloated with indigestible contents) accumulate in cells as lipofuscin granules. Lipofuscin granules are characterized by a single membrane envelope, enclosing yellowish-brown material that can autofluorescence.
Inhibitors of proteases (enzymes that degrade protein) and Vitamin E deficiency result in lipofuscin-like cellular residues — a clue to the origin of lipofuscin. There is evidence that lipofuscin formation inhibits protein degradation, thereby creating a vicious cycle that promotes its own formation [EXPERIMENTAL GERONTOLOGY 36:475-486 (2001)]. In contrast to ceroids — which rapidly accumulate extra− & intra− cellularly in pathologic conditions — lipofuscin accumulates slowly, universally and specifically accumulates in lysosomes [ANNALS OF THE NEW YORK ACADEMY OF SCIENCES; Portas,EA; 959:57-65 (2002)]. The composition of lipofuscin — nearly half protein, one-third carbohydrate and the rest lipid — indicates that it is primarily composed of AGEs rather than lipid peroxidation products [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 236:327-332 (1997)].
Lipofuscin is normally diluted-out of dividing cells, although it is seen in increasing amount in fibroblasts nearing the Hayflick Limit. Lipofuscin accumulation in the non-dividing cells of the brain&heart is very prominent and is, in fact, regarded as a biomarker of aging. Lipofuscin accumulation in retinal pigment epithelial cells may lead to age-related macular degeneration, the leading cause of blindness in the developed world. The fact that lipofuscin accumulates at a higher than normal rate in Alzheimer's Disease and the fact that the disease is also characterized by abnormal tau-protein and amyloid-protein suggests that creation of defective protein and/or problems with removal of defective protein could be the underlying cause of Alzheimer's Disease.
Aging due to free-radicals & glycation of macromolecules other than DNA would be expected more in non-dividing cells than dividing cells — most notably in neurons. That lipofuscin is a component of neuron aging due to free-radical damage is indicated by the high levels of metals (especially iron) in lipofuscin. Oxidative stress has been shown to promote lipofuscin formation, whereas antioxidants reduce lipofuscin formation [FREE RADICAL BIOLOGY & MEDICINE 33(5):611-619 (2002)]. Although antioxidants cannot extend maximum lifespan of organisms as a whole, they may extend the maximum lifespan of neurons or even the entire brain. If so, antioxidants combined with organ replacement could be a means of extending maximum lifespan.
Lysosomes are normally responsible for degradation of aging mitochondria. But as lysosomes become increasingly dysfunctional due to ingestion of indigestible lipofuscin, cells become increasingly populated with aging, swollen mitochondria that produce less energy and more superoxide. Reactive oxygen species produce more aldehydes and more aldehyde-bridges between proteins, resulting in more lipofuscin [EUROPEAN JOURNAL OF BIOCHEMISTRY 269(8):1996-2002 (2002)]. There is thus a positive feedback loop of lipofuscin production, impaired lysosomes, dysfunctional mitochondria and aldehyde formation.
As a cause of death the relative incidence of cancer increases exponentially to age 65 and decreases thereafter. At age 65, 30% of North American deaths are due to cancer, whereas at age 80 only 12% of deaths are due to cancer — mostly because the relative increase of cardiovascular and Alzheimer's Disease is faster than the increase in cancer with age. Nonetheless aging is a major risk factor for cancer, and aging is associated with cancer.
But aging can also be distinguished from cancer, much as with other diseases associated with aging such as atherosclerosis, Alzheimer's Disease, osteoporosis and arthritis. In children, cancers are predominantly leukemias, lymphomas and sarcomas, whereas 80% of adult cancers in the United States are carcinomas. Nearly 90% of mice die of cancer, with about 2/5 of those cancers being lymphomas in males and about 3/5 lymphomas in females. About 30% of male mouse cancers are carcinomas and about 40% are sarcomas [RADIATION RESEARCH; Tanaka,IB; 167(4):417-437 (2007)]. In Werner's Syndrome sarcomas (connective tissue malignancies, usually) are more common than carcinomas. As with the mouse, this may be due to cellular immortalization by ALT rather than telomerase. These patterns do not indicate a simple relationship between aging and cancer.
That there is a distinction between aging and cancer is suggested by the fact that ionizing radiation increases cancer rate, but has less (if any) effect on the rate of aging. Atomic bomb survivors [RADIATION RESEARCH; Preston,DL; 160(4):381-407 (2003) and populations living near a nuclear test site [RADIATION RESEARCH; Bauer,S; 164(4 Pt 1):409-419 (2005)] showed increased noncancer mortality from aging-associated diseases (stroke, heart disease, respiratory disease), but there is no proof that this constituted accelerated aging. Experimental animals subjected to chronic sublethal ionizing radiation (alpha−, beta−, gamma− & X−rays that cause atoms & molecules to form ions) have shown generalize atrophy ("premature aging") and shortened lifespans, but single X−ray & ionizing radiation exposures have more noticeably increased kidney degeneration and cancer (especially leukemia). Other mutagens increase the risk of tumor-formation without reducing maximum lifespan. These results indicate that spontaneous mutations & chromosome breakage are not normal contributors to aging. Mutations due to ionizing radiation are qualitatively different from those occurring "spontaneously" with the passage of time [MUTATION RESEARCH 375:37-52 (1997)]. Mammals with longer lifespans have been shown to have more efficient DNA repair of gamma-radiation [EXPERIMENTAL GERONTOLOGY 37:1203-1205 (2002)]. Nonetheless, the view that ionizing radiation causes accelerated aging is not easily dismissed [AGING; Richardson,RB; 1(11):887-902 (2009)].
Cancer is a disease of DNA, whereas aging is a disease of all organs, tissues, cells and macromolecules. Most cancers are caused by chemical carcinogens, which may result in DNA damage different from DNA damage associated with aging. Cancer is a disease of dividing cells — especially the rapidly dividing cells of the epithelium & blood-forming tissues. Non-dividing cells like neurons or muscle cells don't become cancerous, but aging affects all tissues. A study of 15 rodent species showed that telomerase repression is a feature of large size rather than long life, suggesting that tumor initiation usually occurs during growth and development [AGING CELL; Seluanov,A; 6(1):45-52 (2007)]. Telomerase repression rather than replicative senescence can be the primary anti-cancer mechanism.
DNA must ultimately be responsible for the great variation of maximum lifespan between species. But in this respect DNA (the genome) partly is responsible for the production of reactive oxygen species as well as for the capacity of tissues to withstand oxidative stress & glycation as well as other chemical challenges. If aging is distinguished from cancer by toxin/garbage accumulation and by damage to all macromolecules rather than just DNA, it is nonetheless true that the DNA damage associated with cancer is at least a component of aging. This view is supported by the apparent correlation between maximum lifespan and DNA repair capability seen in species comparisons — as well as by the signs of accelerated aging seen in many DNA repair diseases.
Clues about the molecular mechanisms of aging & cancer in general could be gained by comparative analysis of the mechanisms of segmental progerias leading to specific cancers & specific manifestations of aging. XP, AT & Werner's Syndrome are segmental progerias due to defective NER, defective cell cycle control & defective recombination (respectively) leading to high rates of skin cancer, leukemia & sarcomas (respectively). The cancer symptoms are more prominent with XP & AT, whereas the progeria is more prominent with Werner's Syndrome. Down's Syndrome & Hutchinson-Gilford Syndrome are segmental progerias not particularly associated with high cancer risk. Defective DNA mismatch repair leads to a form of colon cancer (HNPCC) without symptoms of accelerated aging.
What is the relative contribution of reduced vulnerability to cancer due to reduced Insulin-like Growth Factor−1 (IGF−1) to the extended lifespan of dwarf mice and to what extent or by what mechanism is the rate of aging slowed?
Dietary factors, smoking and environmental chemicals can play a significant role in the incidence of cancer, as indicated by the fact that breast cancer in North American women is ten times more common than for women in Japan. And dietary antioxidants — if not supplemental — appear to reduce the risk of cancer. Environmental factors associated with aging or maximum lifespan might cause increased glycation, generalized macromolecule damage and lipofuscin accumulation along with DNA damage.
But in the absence of other diseases, there is a general and exponential increase in the likelihood of contracting cancer as a subject (human or other mammal) ages. There is an increased cumulative effect of DNA mutation and a decline in immune-system function with age. Nonetheless, the pattern of cancer increase associated with aging is very different from immune deficiency disease. Whales have 600 times as many cells as humans yet suffer no greater incidence of cancer. It is improbable that whales have an immune system that is 600 times better than that of humans. Whales must have other special defenses against cancer (which would be well worth learning to understand). The high rate of cancer in rodents is not surprising in light of the proclivity to immortalization associated with their telomeres. But the capacity of mammalian species to detoxify the carcinogenic chemical benzo(a)pyrene to a water-soluble form also correlates well with maximum lifespan [EXPERIMENTAL CELL RESEARCH 116:359-364 (1978)].
DNA damage due to mutagens more readily leads to cancer, but defective DNA repair more readily leads to aging. Nearly all of the "accelerated aging" diseases involve defective DNA repair. Better DNA repair allows the deer mouse to live much longer than the house mouse. It may be that mutagens damage both DNA as well as cellular defenses against DNA damage, but that when DNA repair is defective cells can respond by inducing cellular senescence or apoptosis — preventing cancer, but accelerating aging. With aging the declining efficiency of cellular mechanisms means that there is a decreasing likelihood that cancerous cells will be eliminated by apoptosis.
For technical details about the nature of cancer (and methods of prevention) — see my essay Cancer Death.
Individuals of different species seem to age at different rates for different reasons. Laboratory studies of lifespan is currently only feasible for short-lived species, but if some biomarker could be found for determining biological age (rather than chronological age) then human lifespan studies would be feasible. A biomarker of aging would be a better predictor of life expectancy and future functionality than chronological age. Unfortunately, we even lack a method for biomarker validation. And if a biomarker could be validated for rodents, how could we prove that the biomarker applied equally-well to humans? Without biomarkers of aging we cannot say definitively if "accelerated aging" diseases exist.
Without validated biomarkers of aging, it is difficult to prove that nutrients, drugs or other interventions are slowing aging and extending the maximum lifespans of humans. With biomarkers, it would only be necessary to show reduced deterioration within a reasonable time-frame (a few years) in humans. Without biomarkers, positive proof of an anti-aging intervention for humans could only come by observing effects on lifespan in studies lasting decades or centuries. To be of use within our own lifetimes, the results from short-lived mammals may be the best we can hope for if biomarkers are not found. Despite years of effort, biogerontologists have not had much success in their search for biomarkers of aging [EXPERIMENTAL GERONTOLOGY; Johnson,TE; 41(12):1243-1246 (2006) and BIOLOGICAL CHEMISTRY; Simm,A; 389(3):257-265 (2008)].
Insofar as Caloric Restriction with Adequate Nutrition (CRAN) seems to slow aging in rodents and many other short-lived species, long-term studies of CRAN on monkeys are being conducted to establish if CRAN also slows aging in primates. Although it will take decades for these studies to run to completion and current data is not yet statistically significant, rhesus monkeys on CRAN show the same reductions of body temperature & plasma insulin as CRAN rodents, as well as showing a slower decline in serum DeHydroEpiAndrosterone Sulfate (DHEAS). Men with greater survival in the Baltimore Longitudinal Study of Aging also show reduced body temperature & plasma insulin, along with elevated serum DHEAS — suggesting that these three factors may be biomarkers of biological age [SCIENCE 297:811 (2002)].
Skin biopsies from CRAN & control nonhuman primates have been used to assess glycation & glycoxidation (oxidation of glycation products to form AGEs). Furosine as a measure of glycation increased mildly with age in the control animals and this increase was significantly reduced in CRAN animals. Using pentosine as a measure of glycoxidation, no significant variations were observed — but results for tissues other than skin might have been different [JOURNALS OF GERONOTOLOGY 58A(6):508-516 (2003)].
F2−isoprostanes are stable products of oxidized arachidonic acid which can be readily measured in urine to quantify lipid peroxidation. Plasma concentrations rise dramatically with age in rats, providing support for the association of lipid peroxidation with aging and for the potential of F2−isoprostanes as biomarkers of aging [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 287:254-256 (2001)]. (For details on isoprostanes see Essential Fatty Acids in Cell Membranes.)
A statistically significant trend (with extremely wide variation) of T−cell subsets in mice provides a potential biomarker. Mice with low levels of CD4 & CD8 memory cells, high levels of CD4 naive cells and low levels of P−glycoprotein CD4 cells live 6% longer [JOURNALS OF GERONTOLOGY; Miller,RA; 56A(4):B180-B186 (2001)]. The "biomarker" based on these four T−cell subsets was able to predict longevity of 18-month-old mice with P−values less than 0.003. Mice most often die of cancer, so this might be a better indicator of mortality risk. But if valid, would this biomarker indicate that the immune system theory of aging predominates, or would it indicate that the aging process simply impinges most predictably on the immune system?
With aging the sleeping EEG patterns known as "sleep spindles" and "K−complexes" diminish in number — and it has been suggested that this change can be used as a biomarker of brain aging [CLINICAL NEUROPHYSIOLOGY; Crowley,K; 113(10):1615-1622 (2002)]. There is also reduced circadian signaling with age, and much of this reduction may be due to reduced melatonin secretion [NEUROBIOLOGY OF AGING; Munch,M; 26(9):1307-1319 (2005)].
In the Framingham study (a longitudinal epidemiological study of large size and long duration in Framingham, Massachusetts that has focused on cardiovascular disease risk factors) lung volume (largest volume of air that can be voluntarily expelled from the lung) — which decreases with age in smokers & non-smokers — was well correlated with risk of death in the 45−74 year-old age-range. But even lung volume was inferior to chronological age as a predictor of overall mortality risk. Forced expiratory volume in one second remains the best predictor of all-cause mortality [CHEST; Schunemann,HJ; 118(3):656-664 (2000) and EUROPEAN RESPIRATORY JOURNAL; Young,RP; 30(4):616-622 (2007)], but that does not mean that it is a biomarker of aging.
If mortality risk were the definitive characteristic of aging, then standing in an open field criss-crossed with machine-gun fire would be a biomarker of aging. If aging is damage to organs, tissues, cells and macromolecules then many kinds of damage need to be considered. Certain kinds of damage are more related to specific disease conditions than generalized "aging". Damage to substantia nitra cells leads to Parkinson's Disease, nuclear DNA mutations lead to cancer, glycation of lens crystallins leads to cataracts, etc. Nonetheless, aging increases the predisposition to these disease conditions.
Partly because of the failures to find biomarkers, some biogerontologists question that a unitary process of aging exists — asserting that the phenomenon called "aging" is really multiple degenerative processes operating in parallel. A unitary cause of aging might necessitate discovery of a unitary biomarker. The multiple forms of damage to macromolecules, cells and tissues associated with aging points to multiple causes, and would necessitate multiple biomarkers. Despite the fact that different mechanisms must be involved, the rather uniform slowing of aging seen for dwarf mice and CRAN-diet (versus ad-libitum fed) animals would seem to validate the existence of a unitary aging process — as does the comparison of aging rates between species or breeds of dogs.
But aging can only be the result of damage to macromolecules: proteins, lipids, carbohydrates, and DNA (including telomeres). Causes of aging damage are reactive oxygen & nitrogen species, sugars (glycation), radiation, pathogens, inflammatory cytokines, and accumulated toxins (metals, PCBs, dioxins, etc.). The different aging rates of different species is due to the fact that endogenous damage is produced at different rates (eg, bird mitochondria produce fewer free radicals than mammalian mitochondria), different protective mechanisms exist (eg, naked mole rats arrest cancer growth with contact inhibition), and more long-lived species more effectively eliminate damage (eg with better lysosome enzymes, better DNA repair, better autophagy, etc.). If aging is programmed genetically, it can only be programmed to reduce damage formation or remove/repair damage better or worse.
Exogenous agents can accelerate forms of aging damage. Diabetes and dietary
Advanced Glycation End-products (AGEs) accelerate protein
cross-linking [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Koschinsky,T;
94(12):6474-6479 (1997)]. Immunosenescence is substantially associated with
cytomegalovirus [CURRENT OPINION IN LIPIDOLOGY; Derhovanessian,E; 21(4):440-445
(2009)]. White blood cell telomere attrition is accelerated in obesity and insulin
resistance [CIRCULATION; Gardner,JP; 111(17):2171-2177 (2005)]. There is
considerable overlap in the histopathology of skin photoaging and skin intrinsic
aging [EXPERIMENTAL DERMATOLOGY; El-Domyati,M; 11(5):398-405 (2002)].
A high fat meal elevates plasma inflammatory cytokines more than a high carbohydrate
meal [JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY; Nappo,F; 39(7):1145-1150
(2002)], and plasma inflammatory cytokines are substantially associated with
age-related cataract [AMERICAN JOURNAL OF OPTHALMOLOGY; Klein,BEK; 141(1):116-122 (2006)].
F2−isoprostanes (the best marker of lipid peroxidation) are
substantially elevated in the foam cells of atherosclerotic
plaque [JOURNAL OF CLINICAL INVESTIGATION; Pratico,D; 100(8)2028-2034 (1997)],
and F2−isoprostanes in the urine of smokers drops by
more than one third after two weeks of smoking
cessation [NEW ENGLAND JOURNAL OF MEDICINE; Morrow,JD; 332(18):1198-1203 (1995)].
Because aging is due to multiple forms of damage, there can be no
singular underlying biological age. Cause of death and impairment of
functionality will be a function of which form of damage is the
greatest — which will vary from person to person. Rather than
engage in a fruitless search for a biological age (biomarker of aging),
biogerontologists should seek assays for every possible form of aging
damage. Damage assays can allow for ranking forms of aging damage,
prioritizing interventions, and monitoring intervention effectiveness.
Caloric Restriction with
Adequate Nutrition (CRAN) dramatically extends the maximum lifespan of laboratory animals.
Victims of starvation & malnutrition are not experiencing the life-extending
benefits of CRAN — adequate nutrition (vitamins, minerals, essential
amino acids and essential fatty acids in adequate quantity) is absolutely necessary
for calorie restricted diets to extend lifespan. Because almost every aspect of the
aging process appears to be slowed by CRAN, studying CRAN has become a means of
defining & understanding the aging process itself — including the search for
biomarkers of aging.
Evolutionary biologists have claimed that lifespan increase with CRAN is a physiological
adaptation to increase stress resistance during periods of famine. But lifespan of
Drosophila can be extended by reducing casein or
methionine [NATURE; Flatt,T; 462:989-990 (2009)].
Rats, mice and hamsters experience maximum
lifespan extension from a diet which contains 40−60% of the calories (but all
of the required nutrients) which the animals consume when they can eat
as much as they want. Mean lifespan is increased up to 65% and maximum lifespan
is increased up to 50%, when CRAN is begun just before puberty. Except for the puberty
effect, it is as if all animals are allotted a lifetime supply of food — and those
who eat more slowly live longer because it takes longer to consume all the food.
There is reasonable evidence that the benefits of CRAN seen in rodents apply to humans.
Contrary to former reports of a "J−shaped" relationship between body
weight and human mortality, when corrections are made for smoking and underlying
disease the relationship is linear [NEW ENGLAND JOURNAL OF MEDICINE; Manson,JE;
333(11):677-685 (1995)]. Of course, anorexics who are malnourished for micronutrients
are not examples of human CRAN. There may well simply be a continuum between
CRAN and the metabolic syndrome, meaning the benefits of CRAN are
simply a matter of quantity of calories, versus the idea that CRAN is
some qualitatively distinct metabolic state.
The mechanism by which caloric restriction has such dramatic effects is
unproven, but maturity, thymus shrinkage, DNA-repair decline and tumor
formation is delayed. The experimental animals show more complete oxidation
of fatty acids, with fewer ketones (R'RC=O) in the blood, and cell membranes have less
cholesterol & saturated fatty acids. Collagen cross-linking occurs more
slowly in rats on CRAN which have blood glucose levels reduced about 15%
below controls. Reduction of visceral body fat is associated with reduced
insulin resistance due to
reduced levels of the proinflammatory cytokine [EUROPEAN JOURNAL OF CLINICAL
INVESTIGATION 32(Suppl 3):24-34 (2002)].
Oxidative damage (8−oxodG) to mtDNA is 16 times greater than to nDNA in the livers of
6−month old rats [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Richter,C;
85(17):6465-6467 (1988)]. Although CRAN does not reduce oxidative damage (8−oxodG) to
rat liver nDNA and only reduces oxidative damage to mouse liver nDNA by 19%, it completely
eliminates mtDNA damage in both the rat &
mouse [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Hamilton,ML;
98(18):10469-10474 (2001)]. One year of CRAN in rats has been shown to reduce liver
mitochondrial hydrogen peroxide production from Complex I by 47% [FREE RADICAL
BIOLOGY & MEDICINE; Lopez-Torrez,M; 32(9):882-889 (2002)]. For mammalian species, a
negative exponential correlation has been demonstrated between liver mitochondrial hydrogen
peroxide production and maximum lifespan [JOURNAL OF COMPARATIVE PHYSIOLOGY B;
Perez-Campo,R; 168:149-158 (1998)]. A study of male rats subjected to methionine restriction,
but no restriction of calories, showed the same decreases in mitochondrial reactive oxygen
species and oxidative damage to DNA as was seen with rats subjected to
CRAN [FASEB JOURNAL; Sanz,A; 20(8):1064-1073 (2006)].
Although CRAN animals produce fewer free radicals, their metabolic rate
(oxygen consumption per gram of tissue) is not reduced. The inner mitochondrial membranes
of CRAN animals have a higher saturated/unsaturated fat ratio making them less vulnerable
to proton leak from lipid peroxidation. Both state 3 & state 4 respiration rates
are greatly reduced in brain, heart & kidney tissue [THE INTERNATIONAL JOURNAL
OF BIOCHEMISTRY & CELL BIOLOGY 34:1340-1354 (2002)]. CRAN rats show 15% less plasma
glucose and 50% less plasma insulin than controls, having the same rate of glucose
utilization per unit mass, meaning that glucose is being more efficiently
utilized [JOURNAL OF GERONTOLOGY; Masoro,EJ; 47(6):B202-B208 (1992)].
An ongoing study of rhesus monkeys (which have a maximum lifespan of 40 years)
has shown a 50% reduction in cancer and cardiovascular disease for the animals
subjected to 30% calorie restriction compared to
controls [SCIENCE; Colman,RJ; 325:201-204 (2009)]. Elderly human subjects
(60 years average age) restricting calories 30% for 3 months showed
a 20% increase in verbal memory
scores [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Witte,AV;
106(4):1255-1260 (2009)].
Macroautophagy is normally induced during conditions of starvation. CRAN
in the nematode C. elegans induces macroautophagy, whereas
inhibiting the genes required for macroautophagy inhibits the genes require
for autophagy — and prevents CRAN from extending
lifespan [PLOS GENETICS; Hansen,M; 4(2):e24 (2008)].
Attempts have been made to find a pill that would mimic the effects of CRAN. Using
gene chips it has been found that Metformin results in the same gene expression seen
in CRAN. (See
http://www.lef.org/magazine/mag2003/2003_preprint_bio_01.html
and
http://www.lef.org/magazine/mag2001/sep2001_report_metformin_01.html.) But Metformin only
extends mouse lifespan a third as much as CRAN. By
promoting the growth of insulin receptors, Metformin undoubtedly lowers blood glucose
and reduces extracellular glycation, but free radical production would not diminish.
If aging is ultimately damage to macromolecules, cells and tissues, then any intervention
that slows aging must reduce damaging agents, increase protection against damage or increase
damage repair. There is evidence that CRAN may induce a stress response that reduces fertility,
while increasing defenses such as heat shock proteins, antioxidant enzymes and DNA repair.
Reduced lipotoxicity may also be a factor. But the greatest effect of CRAN in mammals is
probably due to reduced damage by free radicals & reduced glycation, both of which are
associated with more efficient oxidative phosphorylation & glucose utilization.
If these assertions are correct, trying to understand CRAN from the point of view of
changes in cell signalling or gene expression is flawed. For example, it has been reported
that CRAN "down-regulates" apoptosis-related endonuclease in the liver, with the suggestion
that this could be a basis for the life-extending benefits of
CRAN [EXPERIMENTAL GERONTOLOGY; 39(2):195-202 (2004)]. But liver apoptotic-related
endonuclease normally increases with age, and if CRAN slows the aging process, comparisons
with age-matched controls are actually comparing animals of different biological age. To
describe the results of such comparisons as "down-regulation" or "up-regulation" is to
bias the interpretation away from damage and toward cell signalling. Aging cells become
increasingly vulnerable to abberant apoptosis and less capable of initiating apoptosis in
response to cellular defects. The effects of CRAN have been described as increasing
apoptosis in cancer cells while reducing apoptosis in normal cells [MECHANISMS OF
AGING AND DEVELOPMENT; Zhang,Y; 123(4):245-260 (2002)], but a comparison with biologically
matched rather than chronologically matched control animals might show no difference in
apoptosis patterns.
For technical details about CRAN — plus an account of my personal
experiences with CRAN — see my essays
Caloric Restriction with Adequate Nutrition — Overview ,
My Practice of Caloric Restriction with Adequate Nutrition
and My Current Health Regimen — Exercise, Diet, Supplements.
As I stated in the beginning, my primary concern in this essay has been
to elucidate the mechanisms of aging, rather than
methods to prevent it. But the goal
of my understanding is indeed to apply
an understanding of mechanisms to the evaluation of methods. Literature
on methods to extend maximum lifespan which is strongly
grounded in scientific research is rare. The primary reason for this
scarcity is lack of funding for such research.
In contrast with maximum lifespan, there is a vast
amount of literature on ways to extend mean (average)
lifespan through diet & exercise and avoidance of dangers, toxins
& disease. Only 26% of smokers live to age 80, in contrast with
57% of nonsmokers [ADDICTION 97:15-28 (2002)]. A practical
life-extensionist currently has far more to gain by utilizing
information available on extending mean lifespan than by preoccupation
with maximum lifespan. Some misguided life-extensionists have discounted
the use of anti-oxidant supplements because they have only been shown
to be of benefit in extending mean lifespan, not maximum lifespan.
A prime candidate for a biomarker
of aging (which has been a focus of attention in the calorie restriction with
adequate nutrition studies of primates) has been
insulin resistance.
Reduced glycation may be achieved by reduction of typical blood glucose
levels. Low fat meals are one means to achieve this because fatty acids
promote insulin resistance — and greater insulin resistance means that
higher blood glucose levels are required to supply cells with the same
amount of glucose (causing more glycation) [THE NEW ENGLAND JOURNAL OF
MEDICINE 342(19):1440-1441 (2000) and DIABETES CARE 20(11):1774-1780 (1997)].
(Insulin resistance is a fundamental cause of adult-onset diabetes.) Also,
increased consumption of soluble fiber (particularly the beta-glucan found
in oat bran & barley) lowers 24−hour plasma glucose & insulin
concentrations. Experiments demonstrating that lysine-glycation predicts early
death in both CRAN & freely-fed rats makes lysine-glycation a very promising
biomarker candidate [FASEB JOURNAL 14:145-156 (2000)]. (For more on these
subjects see
the metabolic
syndrome)
Although no substance has
been shown conclusively to extend maximum lifespan in humans, a few studies
indicate that some supplements may extend the lives of laboratory mammals
(mice, rats or guinea pigs, usually). The are quite a few studies indicating
that Deprenyl, for example,
has extended the maximum lifespan of a variety of mammals.
There is at least one book (self-published), which is based on a serious
attempt to search the scientific literature for methods to extend
maximum lifespan.
Dr. Thomas Donaldson has reviewed those supplements that appear to extend
the lifespan of mammals in at least one scientific study
in his self-published book A GUIDE TO ANTI-AGING DRUGS. It
would be more accurately titled A GUIDE TO ANTI-AGING SUPPLEMENTS because,
although Procaine, Deanol,
Deprenyl,
Levodopa, Phenformin and Phenytoin
deserve to be called drugs, Vitamin E, Pyridoxine, Pantothenate, Melatonin,
Cysteine, Chromium and Coenzyme Q10 do not. Five mechanisms are identified
by which these supplements work:
(1) anti-oxidation
Dr. Donaldson died early in 2006 and his self-published book
may be difficult to obtain.
Although Hormone Replacement Therapy (HRT) to bring androgens, estrogens and
growth hormone to youthful levels improve cognitive function & muscle tone (among other
benefits) these hormones promote cancer growth and therefore may be dangerous to use until
cancer is preventable & curable. By contrast,
DHEA not only protects against obesity,
diabetes & autoimmune disease, it reduces cancerous tumor-formation [ADVANCES
IN ENZYME REGULATION 26:355-382 (1987)] and can protect against excitotoxic damage in
the hippocampus [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 95(4):1852-1857
(1998)].
The nutraceuticals section of my website describes a number of supplements
which may extend average, if not maximum, lifespan.
Life
Extension Magazine — which is available at no cost online — regularly
publishes articles with many fine citations to research on nutraceuticals
(primarily animal studies) which could potentially be of great benefit in
extending human life.
Cardiovascular function declines and insulin resistance increases
with age for most people. But most of these changes can actually be
attributed to declining physical activity and increasing abdominal
obesity associated with aging — rather than senescence per
se. Higher HDL-cholesterol, lower triglycerides, lower
insulin-resistance & prevalence of diabetes, good cerebral
perfusion, good glucose metabolism, good
cardiovascular function and good endocrine function of older people
who engage in regular vigorous aerobic exercise are nearly to the
level of that seen in their younger counterparts — in sharp
contrast to their sedentary peers [NEW ENGLAND JOURNAL OF MEDICINE
328:533-537 (993); AMERICAN JOURNAL OF PHYSIOLOGY 268:E484-E490
(1995); JOURNAL OF THE AMERICAN GERIATRIC SOCIETY 38:123-128 (1990)].
Health problems caused by sedentary living are too often blamed on
senescence.
Exercise is well-known to lower blood pressure and otherwise improve
cardiovascular health. And, as has been mentioned, exercise can boost
rejuvenating Growth Hormone (GH) more effectively than injections.
Exercise also normalizes other hormone responses to youthful levels
[JOURNALS OF GERONTOLOGY:BIOLOGICAL SCIENCE 51A(1):B30-B37 (1996)] and
reduces insulin resistance [METABOLISM 44(10):1259-1263 (1995)] while improving immune
function [MECHANISMS OF AGING AND DEVELOPMENT 93:215-222 (1997)].
Although excessively strenuous exercise can generate harmful levels of free
radicals, regular endurance exercise protects against free radicals by increasing
muscle levels of SuperOxide Dismutase (SOD), glutathione peroxidase
and reduced glutathione (GSH) (but has no effect on catalase)
[MEDICINE & SCIENCE IN SPORTS & EXERCISE 31(7):987-997 (1999)].
Vitamin E is particularly protective against exercise-induced
free radicals [AMERICAN JOURNAL OF PHYSIOLOGY 264:R992-R998 (1993)].
Vitamin E has a pro-oxidant potential that can only be prevented
by agents like Vitamin C and CoEnzyme Q10, which eliminate the
alpha-Toc. radical [ARTERIOSCLEROSIS, THROMBOSIS
AND VASCULAR BIOLOGY 16:687-696 (1996)]. (For more on exercise, see
Exercise)
According to the Honolulu Heart Program, the best predictors of "successful
aging" were low blood pressure, low blood sugar, abstinence from tobacco and
not being obese. The Framingham study concluded that
by holding 11 different risk factors (such as blood pressure & serum
cholesterol) at the 30-year-old level, women would live to be 97 and
men would live to age 100. As the above review should indicate, many of
the afflictions of aging (including vascular dementia) are the result of
poor cardiovascular health. Therefore, despite the fact that maximum
lifespan is not extended, the effects of extended youth & extended health
would nonetheless be expected from measures extending average lifespan —
cardiovascular health, in particular. Atherosclerosis not only increases
blood pressure and the risk of death from stroke & heart attack, but
reduces the health & function of all organs (including the brain)
through impaired circulation.
It is difficult to
gain much immediate benefit from insights into molecular mechanisms of
aging, but enormous immediate benefit can be gained from reducing
calorie intake (while maintaining adequate nutrition), avoiding tobacco,
avoiding alcohol,
exercising, taking supplements, eating low-fat/high-fiber diets, etc.
Epidemiological evidence indicates that adherence to a vegetarian diet for more
than two decades can increase lifespan 3.6 years [AMERICAN JOURNAL
OF CLINICAL NUTRITION 78(Suppl):526S-532S (2003)].
And cryonics may serve
as "first-aid" to transport us to the time when significant
advances in the elimination of senescence have occurred.
Some geronotologists believe that somatic gene therapy
can accomplish such goals as removing the telomerase gene from somatic cells (to reduce
cancer), migrating mitochondrial DNA into the nucleus and utilizing bird mitochondria genes
to create modified human mitochondria which produce fewer free radicals. With
"adequate funding" these gerontologists believe an ageless
mouse can be created within a decade
(The Methuselah Mouse Prize).
Every year we can add to our lives now increases our chances of
living to the time when technology can eliminate & reverse aging — or
cryonics can induce perfect suspended animation.
This essay is not the place to summarize every practice that can
possibly extend life or delay/avert death. See the pages on this
website dealing with
Health,
Nutraceuticals,
Life-Extension,
CRAN,
Cryonics,
Death by Murder, and my
statistical summary of all causes of death .
A healthy lifestyle, CRAN,
and perhaps even supplements can do no more than slow the aging process or
extend mean lifespan. Enduring youth might be attained if aging could be stopped
at a youthful age, but it seems unlikely that the damage to organs, tissues, cells
and molecules known as aging can be stopped completely. Replacing or repairing
damaged organs, tissues, cells and even molecules seems like a better strategy. These
strategies can restore function to old organisms — can even rejuvenate.
Replacement of old or defective organs is a regenerative technique which has been
tantalizingly close for decades. Only a small fraction of potential candidates for
heart, kidney or liver transplants are able to benefit, because of low availability
and immune incompatibility. The development of a completely mechanical heart remains
out of reach, but there is hope that ventricular assist devices supporting the
left ventricle could benefit most end-stage heart-disease patients. Pigs have many organs
whose size is compatible for human transplant, but immune compatability and the threat
of viral infection remain obstacles.
Although the liver can mostly regenerate lost tissue, wounds to most
body tissues (including myocardial infarction) result in scar formation
rather than regeneration of functional tissue. Stem cells could allow
for true tissue regeneration. Human
Embryonic Stem Cells (ESC) have the
greatest potential to differentiate into any desired tissue type.
Retrovirus
induction of overexpression of certain proteins can generate
induced Pluripotent Stem Cells (iPSC)
from fibroblasts. iPSC are nearly as pluripotent as ESCs. But both
ESCs and iPSCs can form
teratomas
(benign tumors) and induce antigenic tissue rejection (although iPSCs
are less antigenic than ESCs). Antigenicity can be reduced or eliminated
by regenerating the
thymus
gland [NATURE; Chidgey,AP; 453:330 (2008)], by such
means as androgen
blockage> [THE JOURNAL OF IMMUNOLOGY; Sutherland,JS; 175(4):2741-2753 (2005)].
When available, adult stem cells from the target tissue of the afflicted
patient are ideal for avoiding an antigenic response. But too often (as in
cases of tissue degeneration) such stem cells are not available. Stem cells
from the
umbilical cord
cryogenically stored at birth have the potential for tissue regeneration
later in life.
Most attempts at genetic repair have traditionally involved the use of a retrovirus
to insert a new gene into a random position on a chromosome. But by attaching
zinc fingers (which determine
where transcription factors bind) to endonucleases (which break DNA strands) homologous
recombination can be induced to correct and replace defective (or undesired) DNA
sequence. The first applications of this technology are to isolate stem cells from
the bone marrow of patients having blood disease mutations, to correct those
mutations in lab dishes using zinc finger nucleases and to transplant the stem cells
back into the patients [SCIENCE; 310:1894-1896 (2005)].
Regenerative medicine looks for means to mimic salamanders (which can regrow
severed limbs), newts (which can regrow not only limbs, but intestine, jaw and spine)
and zebrafish (which can regrow a heart) — by replacing the dead scar tissue after
a heart attack with new heart cells.
Regenerative medicine uses three different strategies: (1) implantation
of stem cells from culture into an existing tissue structure (2) implantation of
stem cells into a tissue scaffold that guides restoration or (3) induction of
residual cells of a tissue structure to regenerate the necessary body part. A
salamander can not only regenerate a limb, but can regenerate the lens or retina
of an eye and can regenerate an intestine. For regeneration the salamander tissues
form a blastema by dedifferentiation of mesenchymal cells, and the blastema
functions as a self-organizing system to regenerate the
limb [SCIENCE; 310:1919-1923 (2005)]. DNA microarray analysis of salamanders has shown that humoral immune
and local tissue factors control the initial phase of limb regeneration, but nerve-derived
factors later become crucial [BMC BIOLOGY; Monaghan,JR; 7:1-19 (2009)].
The MRL mouse, unlike other mice, can regenerate
damaged heart muscle without scar
formation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Leferovich,JM;
98(17):9830-9835 (2001)]. Regenerative medicine would also aim to replace substantia
nigra cells in Parkinson's Disease and regrow a spinal cord after spinal cord injury.
Multipotent adult progenitor cells, such as bone marrow cells, have been shown to be
capable of replacing myocardial tissue destroyed by ischemic heart
disease [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Orlic,D;
98(18):10344-10349 (2001)].
Senescent cytotoxic T-cells have been removed from the serum of mice by
attachment of iron oxide nanoparticles linked to antibodies and applying
a magnetic field to the serum in an extracorporeal circuit [REJUVENATION
RESEARCH; Rebo,J; 13(2-3):298-300 (2010)].
Injured skeletal muscle has the capacity to regenerate in young mammals, but this
capacity is considerably impaired with aging. Activation & proliferation of muscle
regenerating progenitor cells (satellite cells) is dependent upon signalling from
transmembrane Notch receptors. Notch receptors have several ligands, ie,
extracellular molecules that the receptor requires to function. Upregulation of the
Notch ligand Delta has been shown to be sufficient to restore the regenerative
potential of skeletal muscle in old mice [SCIENCE; Conboy,IM; 302:1575-1577 (2003)],
Caution is advised in upregulating Notch, because overexpression of Notch can lead to
cancer [BREAST CANCER RESEARCH; Dontu,G; 6:R605-R615 (2004)]. The blood
plasma of young mice have been reported to restore the regenerative potential of both
muscle and liver cells in old mice [NATURE; Conboy,IM; 433:760-764 (2005)]. High
levels of TGF−ß (Transforming Growth Factor beta) in the blood of old mice
appears to be the problem. Systemic (serum) TGF−ß is
immunosuppressive [THE JOURNAL OF EXPERIMENTAL MEDICINE; Wahl,SM; 180(5):1587-1590 (1994)]
and aged cells have been shown to produce increased levels of
TGF−ß [IMMUNOLOGY LETTERS; Zhou,D; 36(1):7-12 (1993)]. Muscle regeneration
normally makes use of inflammatory
processes [AMERICAN JOURNAL OF PHYSIOLOGY; Tidball,JG; 288(2):R345-R353 (2005)]
and TGF−ß has been shown to inhibit muscle
regeneration [CIRCULATION RESEARCH; Zhu,S; 94(5):617-625 (2004)].
Organ transplant or even tissue transplant would not be of much benefit for an
aging brain, which is composed of non-dividing, enduring cells (neurons) whose
continued existence is crucial for the retention of knowledge and identity. In this case,
rejuvenation could be done on a molecular level rather than at the tissue or organ level.
For example, Aubrey de Grey has suggested that genes taken from bacteria could be
transmitted into the genome of human neurons to produce enzymes that dissolve &
eliminate lipofuscin, thereby rejuvenating the neuron. The same gene in blood vessel
"foam cells" could reverse atherosclerosis. There is evidence that the
extracellular protein cross-linking due to glycation which
leads to arterial wall stiffening as well as stiffening of the left ventricle can be
reversed by the thiazolium derivative ALT−711, which catalytically breaks
cross-links [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA)
97(6):2809-2813 (2000) and
CIRCULATION; 104(13):1464-1470 (2001)].
Biogerontologist Aubrey de Grey believes that reversing
aging may actually be more feasible than slowing aging, in the same sense that is sometimes
more economical to periodically repair damage than to go to extraordinary expense to
slow the rate of damage. Dr. de Grey believes that the key to rejuvenation is the
repair of seven distinct kinds of damage that represent aging: cell loss, cell senescence,
extracellular protein cross-linking, nuclear DNA mutations, mitochondrial DNA mutations
and the accumulation of garbage inside cells as well as outside cells. He has characterized
the repair of these seven kinds of damage as
Strategies for Engineered Negligible Senescence (SENS).
The seven repair strategies that Dr. de Grey advocates can
be summarized: (1) Cell loss can be repaired (reversed) just by suitable exercise
in the case of muscle, but for other tissues it needs various
growth factors to stimulate cell division, or in some cases it
needs stem cells. (2) Senescent cells
can be removed by activating the immune system against them. Or
they can be destroyed by gene therapy to introduce "suicide genes"
that only kill senescent cells. (3) Protein cross-linking can
largely be reversed by drugs that break the links. But for some of the
links we may need to develop enzymatic methods. (4) Extracellular
garbage can be eliminated by vaccination that gets immune cells
to "eat" the garbage. (5) For intracellular junk we need to
introduce new enzymes, possibly enzymes from soil bacteria, that
can degrade the junk that our own natural enzymes cannot degrade.
(6) For mitochondrial mutations the plan is not to repair them but
to prevent harm from the mutations by putting suitably modified copies of
the mitochondrial genes into the nucleus by gene therapy. The
mitochondrial DNA experiences so much mutation damage because
most free radicals are generated in the mitochondria. If
mitochondrial DNA can be moved into the nucleus it will be
better protected from free radicals, and there will be better
DNA repair when damage occurs. All mitochondrial proteins would
then be imported into the mitochondria. (7) For cancer (the
most lethal consequence of mutations) the strategy is to use gene
therapy to delete the genes for telomerase and to eliminate
telomerase-independent mechanisms of turning normal cells into
"immortal" cancer cells. To compensate for the loss of telomerase
in stem cells we would introduce new stem cells every decade or so.
For more background on Dr. de Grey's approach, see
SENS Overview.
The ultimate rejuvenation, however, will occur further in the future with the advent
of molecular repair technology (nanotechnology) which can fix all kinds of
molecular damage due to aging (as detailed in the book
ENGINES OF CREATION by
K. Eric Drexler).
XXIV. CALORIC RESTRICTION WITH ADEQUATE NUTRITION (CRAN)
XXV. OTHER METHODS TO SLOW AGING
(2) anti-glycation
(3) affecting metabolism
(4) improving the immune system
(5) acting on the brain.
XXVI. REGENERATIVE MEDICINE, STEM CELLS AND REJUVENATION