by Ben Best
Although life-extension practices are of importance, avoiding conventional causes of death — particularly cardiovascular disease, cancer and accidents — should be taken seriously by all life-extensionists whose interest go beyond a fear of cosmetic aging. Cancer is the cause of one-quarter of all deaths in developed countries. Everyone is at risk, but some more than others.
The incidence of cancer increases exponentially with age to age 80. Two thirds of all people with cancer are over age 65. Before the age of 40 cancer strikes fewer than one person in fifty. Between ages 60 to 79 a third of men and a quarter of women develop cancer. Although cancer is rare in children between ages 1 and 14 (fatal disease is rare in children), leukemia is the number one cause of death in that age group. (Leukemia is Latin for "white blood", a reference to the milky appearance of the blood of persons whose white blood cells — leukocytes — have proliferated cancerously.) About half of those diagnosed with cancer are cured and about half are killed by the cancer.
Cancer is a technical subject, but in the second section I attempt to give an overview of cancer without the technical language of cancer or cell biology. Although I use technical language in the technical sections on molecular biology and chemical carcinogenesis, I attempt to define or explain technical terms before I use them. Those who are most interested in the technical sections can still benefit from the background information in the non-technical section. The technical molecular biology sections can be skipped, but some reference to their content will be found in subsequent sections.
Most of the information in this article is a summary of material that is available from textbooks, so I will not bother to provide detailed citations of my sources, except in cases concerning specific or controversial research.
The human body is composed of approximately 200 different cell types that are required to accomplish specific functions. Cells in blood, skin, bone, muscle, brain, etc. must perform distinctive tasks to support the organism as a whole, which supplies them with nutrient, oxygen & waste-removal service. Cancer is a disease of transformed cells. Cancer cells are cells that have reduced their performance of specialized functions and which function more to consume resources of the body while reproducing without limit. If enough cells become cancerous, the organism dies. (Although I am primarily concerned with human cancer, it should be noted that other animals and even plants fall victim to cancer. Laboratory rodents protected from predators & infection usually die of cancer.)
As cancer cells multiply, they form lumps called tumors. (The scientific study of tumors is called oncology, derived from the Greek word onkos, which means "a mass".) Tumors that are benign do not spread to other tissues, may cease to grow and are usually harmless. Skin warts are benign cancer tumors that are caused by viruses. Cancers are malignant when they spread to other tissues. In fact, the term cancer is typically restricted to refer only to malignant tumors. The Greek physician Hippocrates (400 B.C.) named cancer using the Greek word karkinos (crab) because of the creeping, clutching crab-claw appearance of cancerous tissue spreading into other tissue areas.
Methods of categorizing the development of cancer are called staging. The development of a cancer malignancy occurs in three stages:
A single cancerous mutation is not enough to result in a tumor, much less a malignancy. A mutated cell could be shed away (like a skin cell), be killed by the immune system or commit suicide. Even if the mutated cell survives, it will not get beyond the initiation stage if it does not reproduce. However, a single mutated cell may survive for years or decades until some stimulus (a promoter) causes it to reproduce. But if further mutations do not occur to increase survival, reproduction & the ability to spread, the tumor will remain benign and not go beyond the promotion stage. (There is, however, no benign form of leukemia.)
Although cancer is a disease of mutation (DNA damage) almost all cancers are due to environmental factors rather than heredity. Radiation and viruses can mutate DNA, but the predominant initiators of cancer are chemical agents such as aflatoxin, Reactive Oxygen Species (ROS, free radicals) and the polycyclic hydrocarbons found in tobacco smoke and broiled or smoked meat. (Other fried or broiled foods have carcinogens as well, but meats require the most cooking time and have more opportunity to burn.)
Promoters are agents that encourage cell reproduction & growth. Such agents include sex hormones, growth hormone and growth factors (including Interleukin-2, secreted by helper T-cells). At high doses polycyclic hydrocarbons are promoters as well as initiators, by causing enough DNA damage to result in necrotic cell death and consequent proliferation (cell reproduction) to restore the damaged tissues. Cell proliferation also occurs in wound healing and chronic inflammation, which is why tissue irritants act as promoters. Agents that encourage progression to malignancy are those that cause DNA instability. Although most mutations are fatal to cells, a large number of mutations in a large number of cells in a benign tumor has a good chance of producing a few malignant cells.
The susceptibility of different cells, tissues and organs to cancer — and the different manifestations of cancer in different cells, tissues and organs — can be so great that organ-specific cancers have been viewed as completely distinct disease conditions. Mutation (initiation & progression) occurs most readily in rapidly dividing cells, and promotion is unlikely to occur in cells that normally do not divide. For these reasons, cancer is rare in muscle and neurons, wherein cell division is infrequent in adult tissue. Most cancers occur in rapidly dividing cells, namely epithelial stem cells (like skin) or blood stem cells. Cancers also occur most frequently in tissues that interface between the body and the environment (skin, lung and digestive tract). Different organs are subjected to different physical cancer-causing agents. The skin is subjected to ultraviolet light, the lungs are subjected to smoke and the stomach is subjected to burnt, fatty foods. Because cells in different tissues are differentiated differently, different portions of DNA are exposed to the cancer-causing agents.
Normal cells are "good neighbors", which chemically communicate with each other whether to remain static ("contact inhibition") or to grow & multiply (in response to growth factors). Trauma or loss of cell tissue can accelerate cell growth to replace damaged or lost cell tissue. Induction of cell division (mitogenesis) increases mutation because DNA that is replicating is more vulnerable to DNA damage, and because damaged DNA may be replicated before DNA-repair enzymes can repair. Thus promoters are irritating or damaging substances that indirectly foster mutation as a result of chronic mitogenesis. An example of this can be seen in the fact that 80% of cases of gallbladder cancer occur in patients with gallstones.
Malignant cancers spread preferentially from one organ to another. Cancer of the lung commonly spreads to the brain, cancer of the breast spreads to the adrenal glands & lymph nodes, cancer of the colon spreads to the liver and cancer of the prostate spreads to the vertebrae. Bones & lungs are primary targets for cancers originating in other organs.
An estimated 6-10 mutations are necessary for cells to transform to malignant cancer, which is why a high tendency to form mutations is important for the progression stage. Cancer cells reduce DNA repair and reduce cellular defenses against DNA damage. Cancer cells transform cell signalling systems so as to behave as if being constantly stimulated by growth factors. Cancer cells often "immortalize", losing the tendency to become senescent on repeated divisions. Cancer cells develop enzymes that degrade barrier membranes & connective tissue. Cancer cells alter cell surface proteins so as to evade the immune system. Cancer cells acquire motility and the ability to implant into other tissues. (Although fewer than one in two-thousand tumor cells in the blood stream succeed in creating a new colony.) Cancer cells encourage the growth of new blood vessels to nurture their cancerous offspring.
The immune system is a powerful defense against cancer, indicated by the fact that patients taking immune-suppressant drugs for a heart or kidney transplant are five times more likely to develop cancer. Cancer cells displaying abnormal proteins on their cell surface are targets of the immune system. Cancer cells that rid themselves of cell-surface proteins may lose major histocompatibility antigens, making them targets for natural killer cells, which seek-out cells lacking surface proteins. Nonetheless, cancer cells do mutate in ways that elude the immune system.
Most solid tumors can be fully cured only by surgical removal prior to malignant spreading. Malignant cancer can only be treated by agents similar to those that initiate cancer in the first place: DNA-damaging radiation & chemicals. These agents cause the greatest damage to cells that are reproducing most rapidly — the cancer cells. Cancers most vulnerable to radiation & chemotherapy are the cancers with the highest percentage of cells undergoing division at any one time. Side-effect damage is greatest to normal cells that divide rapidly, including blood stem cells, making patients vulnerable to anemia & infection. Patients receiving intensive cancer chemotherapy more often die of infection than of cancer. The risk of developing leukemia secondary to radiation therapy is less than 1% per year subsequent to the therapy. For those who do develop leukemia after radiation therapy, the median time to appearance is ten years.
Tamoxifen is an anti-estrogen used as breast cancer therapy, having the side effects of amenorrhea & hot flashes. Many cancer chemotherapeutic agents have toxic effects on hair follicles, resulting in hair loss that is only partially temporary insofar as hair rarely regrows to full normalcy. Yet despite all the hazards of chemotherapy & radiation treatment, there is a reasonable chance of eradicating the cancer, surviving and eventually recovering from the side effects. Cancers in advanced stages which are described as curable by chemotherapy include testicular cancer, small-cell cancer of the lung, and many forms of leukemia & lymphoma.
Of the 30,000 genes in the human genome coding for different proteins, a few hundred genes regulate growth. Mutations of these genes result in the genetic instability (increased mutability) and uninhibited growth seen in the progressive stage of cancer. The mutated genes are called oncogenes. The normal (pre-mutation) regulatory genes from which they are mutated are called proto-oncogenes. It seems somewhat bizarre to describe normal regulatory genes by referring to them as "pre-diseased", but these genes were discovered by cancer researchers. Developmental biologists had no say in the matter.
Oncogenes (and their corresponding proto-oncogenes) are often named by lower-case Three Letter Acronyms (TLAs) that refer to their tissue and/or cancer-causing virus of discovery. For example, the ras oncogene was found in the rat sarcoma virus. The ras family of oncogenes are more frequently altered in human cancers than any other oncogenes. The src oncogene was found on the Rous sarcoma virus, the myc oncogene was found in the chicken myelocytosis virus, and the erbB oncogene was discovered in chicken erythroblastosis retrovirus. But these genes are part of the genome of all vertebrates, as well as being found in a large number of other species. In fact, src is even part of the yeast genome. Proteins associated with genes are often written with the first letter capitalized (eg, Ras, Src, Myc), but this notation is not consistently followed (eg, ATM, pRb, p53 & WRN proteins and BRCA1, RB, p53 & WRN genes). Human cellular oncogenes may be prefixed with a "c-" (eg, c-Raf) to distinguish them from transforming viral homologs (eg, v-Src).
|
|
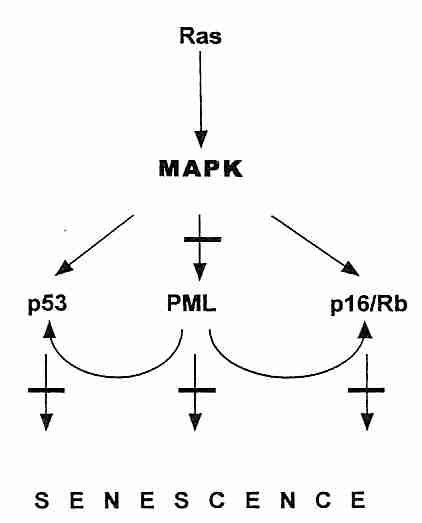
The oncogene most commonly expressed in cancer is ras, which codes for the Ras mitogen receptor which forwards signals to activate cell growth and proliferation through the Raf/MAPK/ERK pathway. Mutations of the ras gene occur in 50% of colon cancers and 90% of pancreatic cancers. Overexpression of ras can also result in stress-induced cell senescence. The second most frequent oncogene in human cancer is myc, which codes for the Myc transcription factor which promotes cell proliferation and induces a catalytic subunit of telomerase, related to the capacity of Myc to immortalize cells. Overexpression of myc can also trigger apoptosis. Apoptosis & senescence are both defenses against cancer, so it is understandable why cells have evolved these defenses in response to ras & myc overexpression.
In addition to oncogenes, the other class of mutations leading to cancer progression are those of the tumor-suppressor genes. These genes typically code for proteins that inhibit cell growth. The Rb gene (mutated in RetinoBlastoma cancer), codes for the pRb protein which controls entry into the S (DNA Synthesis) phase of the cell cycle. The RB gene is recessive, so a person who inherits a defective gene from one parent will experience retinal tumors only if and when the second gene somatically mutates. The p53 tumor-suppressor gene (named for the molecular weight of the 53,000 dalton protein it produces) stops the cell cycle (inducing cellular senescence), but it can also induce cellular suicide (apoptosis) when it detects DNA-damage of such an extent that effective DNA-repair is unlikely. (Apoptosis can be detected by Terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling [TUNEL]). The BRCA1 & BRAC2 tumor-suppressor genes of breast cancer not only affect the cell-cycle, they assist in DNA-damage repair. PTEN (Phosphatase and TENsin homolog deleted on chromosome TEN) tumor-suppressor gene normally prevents the Akt/PI3K survival/proliferation pathway from being overactive. Defective PTEN is found in many cancers. PML is a tumor-suppressor protein that accumulates in the nucleus as aggregates (nuclear bodies), especially at the onset of senescence. PML potentiates p53 action by recruiting p53 to the PML nuclear bodies. The PML nuclear bodies seem to act as a site for fostering regulatory protein interaction in the nucleus, and it is also the site of sirtuin antagonism of p53-induced cellular senescence [THE EMBO JOURNAL; Langley,E; 21(10):2383-2396 (2002)]. At least a dozen tumor-suppressor genes have thus far been identified, but p53 , PTEN and pRb are by far the most important. Senescence activated by p53 is most often due to telomere dysfunction & DNA-damage, whereas senescence activated by pRB is more often due to oncogenes, chromatin disruption or stress.
Although tumor suppressor genes inhibit the cell cycle and oncogenes typically cause excessive growth signalling, cancer cells also require genes to promote angiogenesis (growth of new blood vessels to support new tumor tissue), genes to promote mobility (so cancer can spread) and genes to "immortalize" (allow cells to proliferate indefinitely, without replicative senescence). Telomerase is the usual means by which cells escape the Hayflick Limit and become "immortal". The myc oncogene can induce telomerase expression [GENES & DEVELOPMENT; Wang,J; 12(12):1769-1774 (1996)]. But sometimes cancer cells use a mechanism termed ALT (Alternate Lengthening of Telomeres) — which can be associated with PML protein in the cell nucleus [CANCER RESEARCH; Yeager,TR; 59(17):4175-4179 (1999)]. Although cells that have first become immortalized can usually become cancerous by a ras gene overexpression mutation, the same mutation in normal presenescent cells paradoxically induces cell senescence [CELL; Serrano,M;88(5):593-602 (1997)].
Brain and breast cancers as well as leukemias often have a small portion of tumor cells (as few as 1%) which have stem cell characteristics. These cancer stem cells can be hard to detect, and are resistant to chemotherapy and radiation therapy [ONCOGENE; Lou,H; 26(9):1357-1360 (2007)].
Mutations in oncogenes and tumor-suppressor genes are vastly more probable in cells that deviate from having the correct number of chromosomes (are aneuploid). Genetic instability increases exponentially with aneuploidy. Cervical cancer and non-hereditary colorectal cancer are nearly always characterized by aneuploidy.
It should be noted that mitochondrial gene mutation as well as nuclear gene mutation can play a role in tumor formation. Roughly 11-12% of prostate cancer patients show a defective cytochrome oxidase subunit I mtDNA mutation, which has a mitogenic effect by increasing superoxide production [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Petros,JA; 102(3):719-724 (2005)].
|
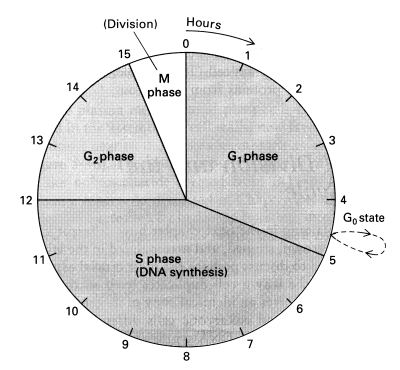
Tumor-suppressor genes act primarily through their influence on the cell cycle. The life cycle of a cell that divides can be described by a four-phase cell cycle composed of M phase, G1 phase, S phase, and G2 phase. M phase is the phase of mitosis, when the cells physically divides. G1 phase is the first Gap phase (or Growth phase), which occurs immediately after mitosis. During G1 the cell is synthesizing proteins and growing to achieve the size a normal cell has before splitting in two during mitosis. S phase is a phase of DNA Synthesis (DNA replication) in preparation for cell division. The G2 phase is characterized by DNA repair of errors introduced during DNA replication, and by preparation for the coming mitosis. Cells that cease dividing — or that rest for extended periods between divisions (such as liver cells) — have temporarily or permanently exited the cell cycle by going into the G0 phase at the end of the G1 phase.
As with most enzyme regulation throughout the cell, enzyme control of the cell cycle is mediated in the nucleus through kinases (enzymes that activate other proteins by the addition of phosphate groups) and phosphatases (enzymes that remove phosphate groups from proteins). The kinases controlling the cell cycle are called Cyclin-Dependent Kinases (cdks), so-called because they cannot act without being conjoined to a cyclin protein. Different cyclins (designated by letters A,B,D,E) are present in different levels during different phases of the cell cycle, being rapidly destroyed by ubiquitin/proteasomes when their services are no longer required. The cyclins were named in the order in which they were discovered, so there is no logical relation between the letter and the cell cycle phase. Similarly, the cdk enzymes are designated by number (cdk4, cdk2, cdk1) according to when they were discovered.
The cyclin D/cdk4 and cyclin E/cdk2 complexes regulate the extremely important and complicated transition between the G1 phase and the S phase. Growth factors induce D & E cyclins (the "start cyclins") leading to cell proliferation, whereas cytokines InterFeroN (IFN-gamma) and Tumor Necrosis Factor (TNF) have the opposite effect. Cyclin D is far more sensitive to growth factors than cyclin E. Overexpression of cyclin E shortens G1, resulting in smaller cells size. The cyclin B/cdk1 complex is active during mitosis (M phase), being activated by dephosphorylation of cdk1 at the end of G2, and deactivated by the phosphorylation (and proteolysis) of cyclin B and rephosphorylation of cdk1 at the beginning of G1. Cyclin B/cdk1 phosphorylates histone H1 for DNA replication, phosphorylates lamins to dissolve the nuclear membrane and phosphorylates vimentin for cytoskeleton reorganization.
|
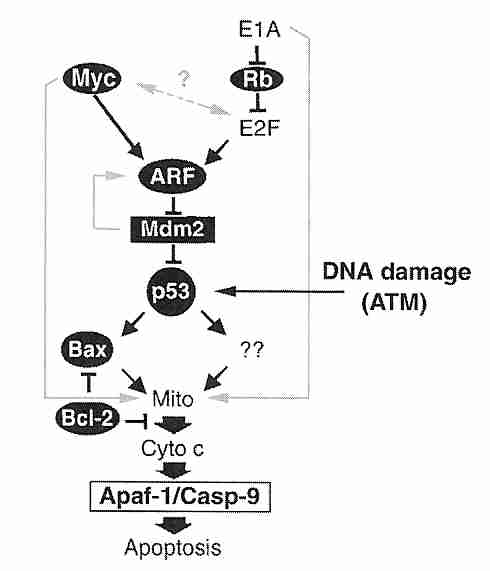
The tumor suppressor protein p53 is a transcription factor that has been called the "guardian of the genome". When DNA damage has occurred, p53 induces the p21 protein to bind with (and inactivate) cdk2, thereby preventing transition from G1 phase to S phase until DNA repair occurs (or causing apoptosis by enhanced if effective repair is not possible). Arrest in G2 phase by p53-induced transcription of the 14-3-3sigma gene. To cause cell death, p53 induces transcription of a number of genes, including those for APF-1 (Apoptosis Protease-activating Factor) and Bax protein. Bax (BAX) protein translocates to the mitochondria where it triggers cytochrome c release, causing the caspase cascade leading to apoptosis. Normally MDM2 oncogene protein binds to p53 preventing its expresssion and promoting p53 ubiquitination (proteasome degradation). MDM2, in turn, is inhibited by ARF protein, which is the target of a number of regulatory proteins acting through E2F protein. ARF sequesters MDM2 in the nucleolus.
Mutations to the p53 gene resulting in non-functional p53 protein occur in more than half of all cancers. Mutations usually involve alterations of critical residues (amino acids) which prevent p53 from binding properly to DNA. In about 10% of cancers p53 is inactivated by overexpression of its inhibitor, the MDM2 oncogene. MDM2 amplification is most often found in cancers of soft tissues (fat, muscle, blood vessels, etc).
Allowing entry into S phase despite the presence of DNA damage results in the higher rate of mutation that is necessary for the progression stage of cancer. Loss of p53 function increases the probability of cellular immortalization. DNA damage due to ionizing radiation activates ATM (Ataxia Telangiectosia Mutated) protein which phosphorylates the chk2/cyclin E complex, which in turn phosphorylates p53 to prevent entry into S phase. An inherited defective ATM gene results in the apoptosis-resistant, cancer-prone disease ataxia telangietasia. ATM may also signal p53 to arrest the cell cycle when telomeres become too short.
Malignancies with p53 mutations, such as ErbB2 in breast cancer, are extremely
resistant to chemotherapy intended to induce apoptosis or cell cycle arrest of cancer
cells. Conversely, malignancies that rarely contain p53 mutations (such as testicular
teratomas) are very sensitive to chemotherapeutic treatment.
|
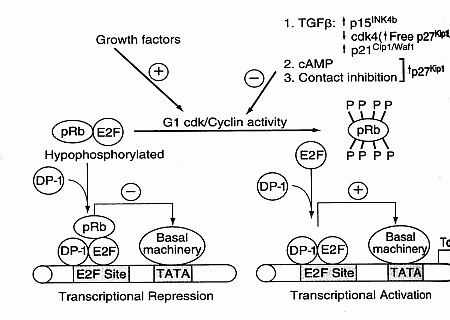
The p16 protein (p16INK4a, INhibitor of CdK4 — which can prevent entry into S phase by binding with cdk4) is also malformed in many cancers. p16 is a major tumor-suppressor protein — increased p16 expression increases cancer resistance [GENES & DEVELOPMENT; Matheu,A; 18(22):2736-2746 (2004)]. The chk4/cyclin D and chk2/cyclin E complexes phosphorylate the pRB protein thereby allowing RNA synthesis from DNA (gene transcription). Proteins p16 and p21 inhibit pRb phosphorylation by cdks, whereas growth factors increase pRb phosphorylation. Transcription factors, such as the E2F protein, are part of the promoter region (along with a Thymine/Adenine-rich area called the TATA box) of inducible genes which provides the start site for RNA transcription from DNA.
E2F transcription factors are required for expression of proteins needed for S phase (DNA Synthesis). E2F genes can also induce transcription of hTERT (telomerase reverse transcriptase), resulting in telomere synthesis. When pRB is not phosphorylated it binds to E2F, thereby preventing gene transcription. Thus, the phosphorylation state of pRB provides another means of controlling the G1/S checkpoint. Many cancers have mutations in the RB gene leading to malformed pRB proteins. The protooncogene protein Bcl-2 also affects the G1/S checkpoint by blocking E2F. Again, by allowing easy entry into the S phase, defective pRB, Bcl-2 & p16 proteins increase the rate of mutation.
Contact inhibition is a mechanism for arresting cell growth in collections of cells. Cancer has never been observed in naked mole rats, which are hypersensitive to contact inhibition [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Selvanov,A; 106(46):19352-19357 (2009)].
The G1 phase cyclins D & E can be induced by growth factors from outside the cell and can be suppressed by antiproliferation cytokines like Interferon-gamma (IFN-gamma) and Tumor Necrosis Factor-alpha (TNF-α). Cell growth can be triggered by the ERK (Extracellularly Regulated Kinase) subfamily of the MAP (Mitogen-Activated Protein) subfamily of Kinases (MAPK). Mitogens (substances outside the cell which stimulate cell proliferation) and growth factors usually act on cell surface receptors to activate signalling molecules which affect gene transcription leading to accelerated cell growth. Oncogenes are generally genes that amplify or otherwise affect the mitogenic/growth factor cell signalling from cell surface receptors and/or transcription factors associated with rousing cells from G0 phase and/or increasing growth rate. Oncogene proteins are also present in the intracellular signalling from integrins (surface proteins mediating cell motility, attachment and associated growth signals). Whereas tumor suppressor gene mutations can be described as "faulty brakes", oncogenes can be characterized as "stuck accelerator pedal". Both accelerated cell-cycle speed and lack of control over the transition between G1 phase and S phase lead to the increased mutation rate as well as increased cell proliferation associated with the progression stage of cancer (although oncogenes often are initiators as well as promoters).
Most growth factors act by binding to transmembrane tyrosine kinase receptors on the cell surface membrane. Such growth factors include EGF (Epithelial Growth Factor), VEGF (Vascular Endothelial Growth Factor), PDGF (Platelet-Derived Growth Factor), insulin, IGF (Insulin-like Growth Factor), and many others. From the point of view of cancer, the most notable cell signalling pathway is the one activated by the Ras protein attached to the inner cell surface associated with the tyrosine kinase receptor.
The Ras protein activates a cascade (the Ras/Raf/MEK/ERK pathway) including MAPK (Mitogen-Activated Protein Kinase), which ultimately activates the gene transcription factor family AP-1 (Activator Protein-1, usually composed of a Fos protein joined to a Jun protein) resulting in increased gene transcription of cyclin proteins that drive the cell cycle. Oncogenes of the Ras protein (mutated proto-oncogenes) cause Ras to be perpetually active, whether a growth factor is present or at the tyrosine kinase receptor or not. Nearly a third of all human cancers have the ras oncogene, forming a defective Ras protein. Both the p53 tumor-suppressor gene and the ras oncogene promote VEGF secretion — promoting angiogenesis (growth of new blood vessels required to support new, rapidly-growing cancer tissue).
| Cell Receptor Signalling to the Nuclear Transcription Factors |
|---|
![[Cell Receptor Signalling to the Nuclear Transcription Factors ]](signals.gif)
|
The transcription factor proteins Fos, Jun and Myc come from proto-oncogenes designated as immediate early genes because they are expressed rapidly after mitogen stimulation and have a short half-life. (Restated: The proteins from immediate early genes are transcription factors produced in response to cell stimuli, and these transcription factors cause the production of functional proteins.) The immediate early gene proteins cause transcription of delayed response genes which produce proteins such as E2F, cyclin D and cyclin E, which increase cell cycle activity and growth. Oncogenes produced mutated forms of the immediate early genes which can increase the activity of the proteins they form.
Overexpression of the Myc growth factor can lead to the angiogenesis and tumor-cell invasiveness that characterizes malignancy. Myc overexpression increases Bcl2 protein and decreases p53 protein, both effects being anti-apoptotic and pro-proliferative. Bcl2 prevents Bax protein from releasing cytochrome c from mitochondria. Cytochrome c release triggers apoptotic activation of caspase proteolytic enzymes. Burkitt lymphoma is characterized by Myc/Bcl2 overexpression and apoptosis inhibition. Myc shows a special affinity for CpG islands, and its DNA binding is significantly inhibited by DNA methylation. Myc DNA targets are often tumor-suppressor genes that are frequently silenced by methylation in cancer. Myc enhances histone acetylation [GENES & DEVELOPMENT; Fernandez,PC; 17(9):1115-1129 (2003)].
G protein linked receptors for epinephrine, serotonin & chemotactic cytokines wind through the cell surface membrane like a serpent in seven transmembrane segments. The G protein linked receptors produce intracellular second-messengers such as cyclic AMP (cAMP), cyclic GMP (cGMP) and calcium, which can activate transcription factors such as CREB & NF kappaB, as well as AP-1. The ability of G-protein-coupled signaling pathways to undergo desensitization to receptor substances ("tolerance") is part of the basis for addiction to opiates, caffeine, benzodiazepines and nicotine. Mutations resulting in chronic activation of G protein receptors are found in some cancers.
| The Akt/PI3K Pathway |
|---|
![[ The Akt/PI3K Pathway ]](Akt_PI3K.jpg)
|
PTEN tumor suppressor protein normally acts primarily by dampening the PI3K/Akt pathway that promotes cell growth and inhibits apoptosis [MEDICAL SCIENCE MONITOR; Chu,EC; 10(10):RA235-RA241 (2004)], in contrast to TNF (Tumor Necrosis Factor) which activates Akt/PI3K signalling [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Ozes,ON; 98(8):4640-4645 (2001)]. Secondarily PTEN dampens the Raf/MAPK/ERK cell proliferation pathway [HUMAN MOLECULAR GENETICS; Weng,L; 11(15):1687-1696 (2002)]. Thus, PTEN mutations which make the protein defective release two pathways which can lead to excess proliferation and thus to cancer. PTEN also has lipid phosphatase activity which antagonizes the inhibitory effect of the PI3K/Akt pathway on autophagic cell death (an alternative to apoptosis as a means of "cell suicide" )[JOURNAL OF BIOLOGICAL CHEMISTRY; Arico,S; 276(38):35243-35246 (2001)].
A primary target of the PI3K/Akt and Ras pathways is the kinase mTOR (m Target Of Rapamycin). The mTOR kinase signals cells to grow and proliferate, particularly promoting formation of new blood vessels required by rapidly-growing tumors [NATURE; Shaw,RJ; 441:424-430 (2006) and NATURE; Bernardi,R; 442:779-785 (2006)]. Rapamycin is an anti-cancer drug that inhibits mTOR, and is particular effective in cancers involving PTEN loss of function [ANNALS OF ONCOLOGY; Vignot,S; 16(4):525-537 (2005)]. Mice fed rapamycin have shown reduced cancer and extended lifespan [NATURE; Harrison,DE; 460:392-396 (2009)].
Healthy wound-healing involves a well-coordinated immune/inflammatory response. Neutrophils and macrophages (immune system cells) enter the wound and fight bacteria by creating toxic free radicals like hydrogen peroxide, peroxynitrite and the hydroxyl radical. Antioxidant enzymes like selenium-containing glutathione peroxidase and thioredoxin reductase protect neutrophils, macrophages and other tissues from the free radicals intended to destroy pathogens. Macrophages release growth factors to promote tissue re-growth.
With chronic inflammation, however, these natural mechanisms run amuck into cycles of tissue regeneration and destruction, creating an environment conducive to cancer-development [NATURE; Coussens,LM; 420:860-867 (2002) and NATURE REVIEWS, IMMUNOLOGY; Karin,M; 5(10):749-759 (2005)]. An estimated 20% of all cancers are due to chronic inflammation. Continuous exposure to the free radical peroxynitrite leads to DNA mutation. Growth factors from macrophages promote proliferation of new cancer cells. An estimated 15% of cancers are attributed to inflammation associated with chronic infections, such as hepatitis, papillomavirus, and the gastric bacterium Heliobacter pylori [JOURNAL OF INTERNAL MEDICINE; Kuper,H; 248(3):171-183 (2000)]. Non-infectious causes of chronic inflammation such as tobacco smoke and asbestos also contribute significantly to cancer.
Nuclear Factor−kappaB (NF−κB) is a transcription factor that has crucial roles in inflammation, immunity, cell proliferation and apoptosis — all of which play a role in cancer causation [ANNALS OF THE NEW YORK ACADEMY OF SCIENCES; Delhalle,S; 1030:1-13 (2004)]. NF−κB is normally bound to IκB protein in the cytoplasm, but is released to enter the nucleus when infection, oxidative stress or pro-inflammatory cytokines (like TNF−α, Tumor Necrosis Factor−alpha and IL−1 — InterLeukin−1) cause ubiquitination and subsequent protease degradation of IκB. NF−κB increases transcription of genes coding for TNF−α and IL−1, which can result in a positive feedback loop. Aberrant NF−κB activity is often found in cancer and chronic inflammatory disease. The antioxidant nutraceutical N−AcetylCysteine (NAC) is particularly effective at blocking TNF−α activity, because of its ability to interfere with TNF−α binding to its receptor [THE EMBO JOURNAL; Hayakawa,M; 22(13):3356-3366 (2003)].
Although the main carcinogenic effects of inflammation are on tumor promotion and progression, chronic inflammation can also lead to tumor initiation. NF−κB induces the expression of anti-oxidant genes (such as for mitochondrial superoxide dismutase), but it also causes induction of inducible Nitric Oxide Synthetase (iNOS), leading to DNA damage (increasing cancer initiation) while at the same time inhibiting apoptosis (inhibiting the elimination of cancer cells) [NATURE REVIEWS, IMMUNOLOGY; Karin,M; 5:749-759 (2005)]. The endotoxin LipoPolySacccharide (LPS) (derived from the outer membrane of Gram-negative bacteria) contributes to septic shock by its ability to activate NF−κB to induce iNOS [JOURNAL OF BIOLOGICAL CHEMISTRY; Xie,Q; 269(7):4705-4708(1994)]. (Septic shock is the main cause of multiple organ system failure, the leading cause of mortality in intensive care units.) During infection DNA damage in pathogens is beneficial, but in chronic inflammation the body's own cells become the victims. Moreover, peroxynitrite is a powerful inducer of pro-inflammatory cytokine release, which also creates conditions for positive feedback [JOURNAL OF BIOLOGICAL CHEMISTRY; Matata,BM; 272(3):2330-2335 (2002)].
Experimental elimination of NF-κB in mice suffering from chronic inflammation has resulted in apoptosis of premalignant cells and a halting of tumor progression — suggesting that NF-κB is the link between inflammation and cancer [NATURE; Covert,MW; 431:461-466 (2004)].
The hyperproliferation associated with chronic inflammation leads not only to decreased genome stability and increased mutation, but to hypermethylation (see Section F).
The fact that 30% of rodents have had cancer by age 3 and 30% of humans have not had cancer until age 85 indicates a profound influence of genetics upon cancer susceptibility. There are also great differences in susceptibility to cancer between different organs and tissues, not all of which can be attributed to environmental differences. Cancer results from DNA changes that re-direct cellular activity away from anything other than reproduction, growth and invasion of other tissues.
Although cancer is a disease of DNA mutation, the vast majority of those mutations are acquired due to environmental influences (especially chemical) rather than by heredity. It is, however, possible to inherit a predisposition to cancer which, nonetheless, requires additional environmental influences to become manifest. Inherited cancer mutation are most often recessive tumor suppressor genes such as BRCA2 (breast cancer), APC (colon cancer), RB (retinoblastoma) or p53 (breast cancer). Were the gene not recessive, the embryo would quickly be consumed by cancer. Being recessive means that an additional mutation is required for the genes to become manifest. But by already having one of the two mutations required for activity, the homozygous mutant conditions necessary for malfunctioning tumor suppressor protein becomes much more likely to occur.
Almost all cancer-causing viruses are DNA rather than RNA viruses. Although oncogenes were discovered in viruses (which took them from animal DNA), less than 15% of human cancers are due to viruses (mostly in underdeveloped countries). The oncogenic DNA viruses consist of four main groups: papoviruses, hepadnaviruses, herpesviruses and adenoviruses. Although adenoviruses can cause tumors in rodents, there is no evidence that they do so in humans.
The Human PapillomaVirus (HPV) associated with cervical carcinoma is a papovirus that produces two oncoproteins, one of which binds to p53 protein and the other of which disrupts the action of the RB gene. Insofar as both p53 and pRb are both tumor suppressor proteins that can stop the cell cycle, blocking the action of both proteins is a major step leading to cellular "immortalization" through telomerase (usually), or ALT (Alternative Lengthening of Telomeres). The virus is most effectively carcinogenic when the HPV DNA becomes incorporated into the host cell DNA.
Hepadnaviruses infect the liver and thereby cause a large portion of the cancers seen in underdeveloped countries. The most notable member of this virus family is hepatitis B, which produces a protein that affects the myc proto-oncogene and activates the transcription factor NF kappaB. Hepatitis C, an RNA virus, is also associated with liver cancer, but the carcinogenic effect is probably secondary to the proliferation associated with chronic infection.
Herpes simplex virus probably contributes urogenital and oropharangeal cancers. In developed countries the Epstein-Barr herpesvirus is associated with mononucleosis, a non-malignant form of lymphocyte proliferation. But in wet sections of Africa Epstein-Barr causes Burkitt's lymphoma, the predominant cancer in the children of that region. And in southern China Epstein-Barr is responsible for many cases of nasopharyngeal carcinoma.
Although retroviruses and other RNA viruses contribute to cancer in animals, they rarely do so in humans except through indirect effects. The HIV retrovirus is associated with cancer in AIDS patients, but most often this effect is due to reduced immune system action against herpesviruses.
Epigenetic refers to modifications of gene expression that occur without
changes in DNA sequence. Many such changes may be heritable. The primary mechanisms of
epigenetics are histone acetylation (causing gene expression), histone deacetylation
(causing gene silencing), histone methylation and DNA methylation (both usually causing gene
silencing). Histones are DNA-associated proteins that control the compacting of DNA
into chromatin. Acetylation of H3 & H4 histones exposes associated
DNA for possible transcription, whereas deacetylation of those histones results in
chromosome compaction, making DNA less accessible. By making DNA more accessible, histone
acetylation can facilitate DNA repair, particularly Nucleotide Excision Repair (NER), while
at the same time increasing vulnerability to DNA damage.
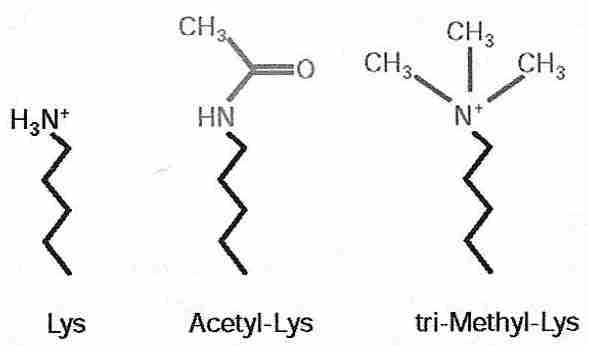
|
Histone acetylases and histone deacetylases control DNA compacting by the addition or removal of acetyl groups to the lysine residues of histones. Histone acetylase addition of an acetyl group to the lysine amine group both nullifies the positive charge on the nitrogen and makes the group more hydrophilic (because the carbonyl group can hydrogen-bond). Because DNA is nucleophilic (cation-attracting), acetylated lysine is less attracted to the DNA and (helped by the increased hydrophilicity) histones are less attracted to the DNA — resulting in the DNA becoming less compacted and more exposed for DNA (gene) expression. Conversely, histone deacetylase reduces hydrophilicity and increases electrophilicity (anion-attraction) — resulting in DNA becoming more compacted and silencing gene expression.
Methylation of histones is usually less reversible than acetylation and is thought to be a mechanism for heritable gene silencing [CELL 109:801-806 (2002)]. Histone methylation of lysine causes gene silencing by making lysine more hydrophobic and more electrophilic (the methylated nitrogen cation is more positively charged) thereby increasing compaction and silencing DNA expression. Methylation enzymes can add one, two or three methyl groups to lysine. Arginine can also be mono- or di-methylated to increase gene silencing. If dimethylation enzymes exist, they are far less active than methylation enzymes.
(For more information on histones and DNA repair, see DNA Damage an DNA Repair. For more information on gene silencing by deacetylation, see Sirtuins and Deacelation. )
Methylation of DNA — specifically the addition of a methyl group to
carbon 5 of cytosine — is more reversible than histone methylation and is usually
associated with gene silencing, but may sometimes increase gene expression. (DNA is composed
of four nucleobases:cytosine, guanine, adenine, and thymine, whereas in RNA
uracil is used instead of thymine.)
| PYRIMIDINES | PURINES |
|---|---|
![[ PYRIMIDINES ]](pyrimidines.gif)
|
![[ PURINES ]](purines.gif)
|
|
A cytosine adjacent to guanine nucleotide in the 5'-direction forms a so-called CpG dinucleotide (5'-CG-3', with "p" referring to the phosphate group linking the two nucleobases). Cytosines in CpG dinucleotides are readily methylated. Because 5-methylcytosine can be deaminated to thymine (a mutation), CpG dinucleotides are not common in the genome [BLOOD; Singal,R; 93(12):4059-4070 (1999)]. Sequences rich in CpG (so-called CpG islands) are almost always (if not always) found in promotors of housekeeping genes that are frequently switched-on. How methylation of promoter CpG islands is normally prevented is the subject of much speculation. Possibly poly(ADP-ribosyl)ation plays a role [THE FASEB JOURNAL; Zardo,G; 16(10):1319-1321 (2002)], or gene sequences which provide structural protection [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Feltus,FA; 100(21):12253-12258 (2003)].
In cancer, there is a general demethylation of the whole genome, whereas hypermethylation
of CpG islands occurs in the promoters of tumor suppressor genes in many
cancers [NATURE GENETICS 21:163-167 (1999)]. The metal nickel is a known carcinogen which
is not mutagenic, but which induces CpG island
hypermethylation [MOLECULAR AND CELLULAR BIOLOGY; Lee,Y; 15(5):2547-2557 (1995)]. Promoter
hypermethylation is such an early even in ovarian cancer that hypermethylation of serum
promoter DNA may be a means of early
detection [CANCER RESEARCH; de Caceres,II; 64(18):6476-6481 (2004)]. Promoter CpG
island methylation normally increases incrementally with aging in colon cells, but moreso
in colorectal cancer (associated with diminished MMR DNA repair) leading to the claim that colorectal cancer is a kind
of accelerated
aging [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Toyota,M;
96(15):8681-8686 (1999)].
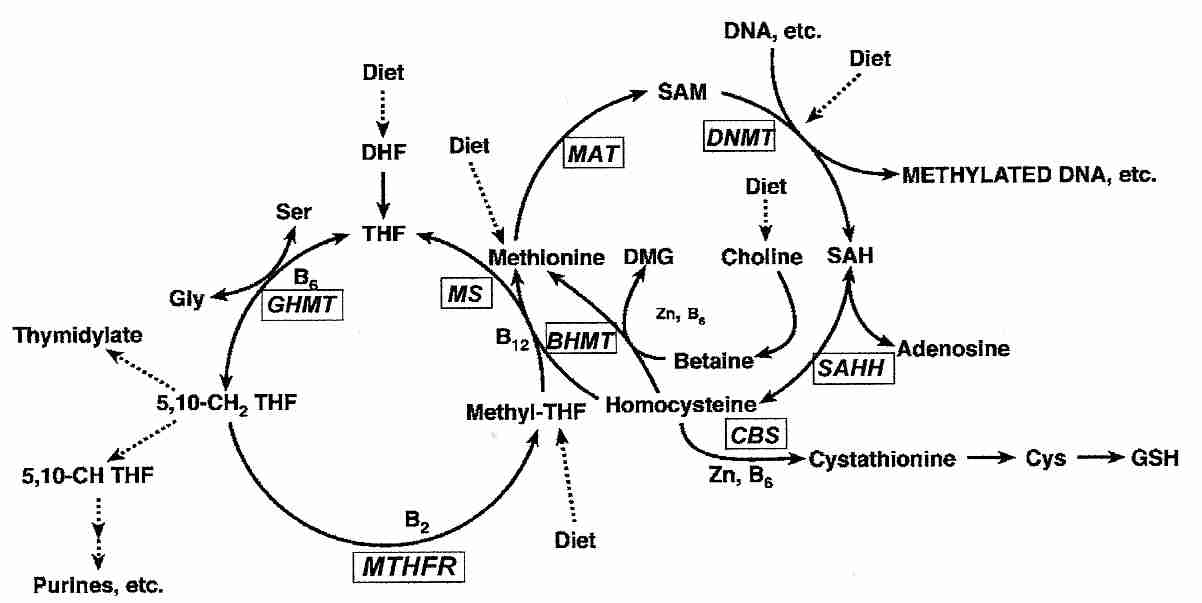
|
Methyl groups required for DNA synthesis & methylation ultimately come from folic acid (THF, TetraHydroFolate) in the diet, which is converted to the methyl donor S−AdenosylMethionine (SAM). The enzyme DNA cytosine-5-MethylTransferase 1 (DNMT1) catalyzes the methylation of DNA by SAM. During DNA replication DNMT1 copies parental DNA methylation patterns by being considerably more active in regions where one DNA strand is methylated [CELL RESEARCH; Zardo,G; 15(9):679-690 (2005)]. Hypermethylation associated with hyperproliferation is likely an important factor in the association between inflammation and cancer [CANCER RESEARCH; Issa,JJ; 61(9):3573-3577 (2001)].
Hypomethylation is also carcinogenic, which is why dietary folic acid deficiency is associated with increase susceptibility to cancer [THE JOURNAL OF NUTRITION; Jacob,RA; 128(7):1204-1212 (1998)]. Acetaldehyde from alcohol metabolism degrades folic acid, which may explain some of the relationship between alcohol consumption and breast cancer [CARCINOGENESIS; Freudeheim,JL; 26(6):931-939 (2004)] as well as colon cancer [THE JOURNAL OF NUTRITION; Choi,S; 129(11):1945-1950 (1999)].
The DNA repair enzyme O6-MethylGuanine-DNA MethylTransferase (MGMT) is frequently repressed by hypermethylation in colon cancer, which thereby allows alkylating agents to cause the G:C-to-A:T conversions which are behind the K-ras mutation seen in about half of colorectal carcinomas. Conversely, lack of MGMT hypermethylation in breast cancer explains the fact that K-ras mutations are rarely seen in breast carcinoma [WORLD JOURNAL OF GASTROENTEROLOGY; Qi,J; 11(13):2022-2025 (2005)].
An assay of 15 major tumor types found a high incidence of hypermethylation in promoter-associated CpG islands for tumor suppressor genes (p14ARF, p16INK4a), DNA repair (MGMT, BRCA1) and metastasis (cell adherence, TIMP3), for example. MGMT promoter hypermethylation was found in 39% of colon cancers and 34% of brain cancers. Promoter hypermethylation of p16INK4a was found in 48% of lymphomas and 39% of pancreatic cancers. Kidney tumors displayed 78% TIMP3 promoter hypermethylation. For a complete table, see [CANCER RESEARCH; Esteller,M; 61(8):3225-3229 (2001)].
Many cancer-causing chemicals block DNA methylation. Dietary deficiency of nutrients required for methylation (methionine, choline, folate and Vitamin B12) lowers liver DNA methylation and increases liver cancer in experimental animals [JOURNAL OF NUTRITIONAL BIOCHEMISTRY 4:672-680 (1993)].
The first carcinogenic (cancer-causing) chemicals discovered — and the first to
be used in chemotherapy
(to cause mutations in leukemia cancer cells) — were the nitrogen mustards. Nitrogen
mustards and nitrosoureas are known as alkylating agent carcinogens because they can
attach alkyl groups to DNA, RNA or proteins. Alkylated DNA bases frequently cause mispairings
during replication. For example, O6 methylguanine pairs with thymine instead
of with cysteine, leading to GC-to-AT mutation. The two chloroethyl chains of nitrogen mustard
can substitute their chlorines for covalent bonds with electronegative DNA groups on each
strand of a chromosome. When DNA strands are locked together at several sites, the strands
cannot separate properly during DNA replication and the cell dies. Alkyltransferase
DNA repair
enzymes directly fix alkylation damage by transferring an alkyl (methyl or ethyl) to a
cysteine acceptor site on the alkyltransferase.
| Nitrogen Mustard | Alkylation of Two Nucleic Acid Bases by Nitrogen Mustard |
|---|---|
![[Nitrogen Mustard]](mustard.gif)
|
![[Alkylation of Two Nucleic Acid Bases by Nitrogen Mustard]](alkylate.gif)
|
Most carcinogenic chemicals are electrophiles (molecules that can accept a pair
of electrons) that form covalent bonds with nucleophiles (electron-rich molecules
that may or may not have a negative charge). Electrophilic carcinogens target the sulphur,
oxygen and nitrogen groups that make the amino acids cysteine, tyrosine and histidine
(respectively) nucleophilic. The DNA phosphodiester backbone is nucleophilic. The purine
& pyrimidine nitrogens & oxygens are very nucleophilic, with guanine being the
nucleic acid that reacts most avidly with electrophiles.
| PYRIMIDINES | PURINES |
|---|---|
|
|
|
The most common chemical carcinogens are polycyclic hydrocarbons that are generated by burning carbon-containing materials such as tobacco, meat, fried food (especially when the same oil is used repeatedly) or wood. Unlike alkylating agents, the polycyclic hydrocarbons and most other so-called carcinogenic chemicals are actually innocuous precarcinogens that must undergo a metabolic conversion before becoming carcinogenic electrophiles. Polycyclic hydrocarbons are hydrophobic, so the liver must add hydroxyl groups to make them water-soluble enough to be excreted in the urine. This is the reason that the Ames Test for determining the carcinogenic potential of chemicals using bacteria requires the addition of mammalian liver cell extracts.
The elimination of precarcinogens is typically a two-stage reaction involving two different sets of enzymes, designated phase 1 and phase 2 enzymes. Phase 1 enzymes perform oxidations or reductions on the hydrophobic precarcinogens preparing them for conjugation reactions by phase 2 enzymes (such as epoxide hydrolase, which converts an epoxide to non-reactive -OH groups). Phase 2 enzymes produce non-toxic, water-soluble compounds that are readily excreted in the urine. The phase 2 detoxifying enzyme glutathione-S-transferase, for example, increases the water solubility of electrophilic, lipophilic toxins by addition of the tripeptide reduced glutathione (GSH). Unfortunately, phase 1 enzymes can often produce highly carcinogenic electrophiles that are not detoxified by phase 2 enzymes.
Phase 2 enzymes not only include those that conjugate electrophilic toxins for elimination, but also include the general antioxidant defense enzymes normally present in animal cells. Phase 2 enzyme inducers such as the protein Nrf2 attach to a region of DNA known as the Antioxidant Response Element (ARE) to induce transcription of factors that increase synthesis of detoxification (conjugation) enzymes, antioxidant enzymes and glutathione.
Normally the transcription factor Nrf2 is present in the cytoplasm attached to a Keap 1 protein. But when cells are stressed by free radicals or toxic electrophiles, Keap 1 releases Nrf2 allowing Nrf2 to enter the nucleus and join to a MafK protein which is needed for Nrf2 attachment to the ARE. Signals from the PI3K/PKC pathway and the Raf/MEK/ERK pathway can accelerate the release of Nrf2 and the influence of Nrf2 on ARE transcription [THE JOURNAL OF TOXICOLOGICAL SCIENCES; Numazawa,S; 29(2):81-89 (2004)]. NADPH oxidase enzyme in cell membranes controls the ERK pathway signalling [JOURNAL OF BIOLOGICAL CHEMISTRY; Papaiahgari,S; 279(40):42302-42312 (2004)].
The phase 1 cytochrome P450 enzyme CYP1A1 (aryl hydrocarbon hydroxylase) can be induced to higher levels by the polycyclic hydrocarbons in cigarette smoke. Smokers with a genetic defect resulting in greater quantities of CYP1A1 are more likely to get lung cancer. Many isothiocyanate phytochemicals from cruciferous vegetables are potent phase 2 enzyme inducers, but some are phase 1 enzyme inducers as well — allowing for rapid detoxification and removal of carcinogens [JOURNAL OF NUTRITION; Talalay,P; 131(11):3027S-3033S (2001)].
Benzo(a)pyrene is a prototypic polycyclic hydrocarbon precarcinogen.
Cytochrome P450 phase 1 enzymes in liver cells can add hydroxyl groups to
the molecule, but only by forming an epoxide intermediate. If the epoxide forms in the
K region of benzo(a)pyrene, the phase 2 enzyme epoxide hydratase can
easily metabolize it. But epoxide hydratase cannot so easily metabolize an epoxide near
the Bay region. Epoxides
are highly reactive electrophilic groups that can attach to DNA forming bulky adducts
that block DNA synthesis, resulting in noncoding lesions.
| Benzo(a)pyrene | Epoxide Formation Near the Bay Region |
|---|---|
![[Benzo(a)pyrene]](BenzoPyr.gif)
|
![[Epoxide Formation Near the Bay Region]](BenzPRxn.gif)
|
Aflatoxin B1 is a very potent carcinogen produced by a
mold found on many foods in underdeveloped countries — and is a frequent cause
of liver cancer in those countries. Aflatoxin forms an epoxide which forms a
covalent bond primarily with the N-7 nitrogen of guanine. Insertion of flat planer
rings of polycyclic hydrocarbons into DNA can distort the helix, leading to
frame-shift mutations (ie, insertion or deletion of bases) during DNA replication.
| Aflatoxin B1 | Aflatoxin B1 forming Epoxide |
|---|---|
![[Aflatoxin B<SUB>1</SUB>]](Aflatoxin.gif)
|
![[Aflatoxin B<SUB>1</SUB> forming Epoxide]](AflatRxn.gif)
|
Many nitrosamines (compounds of the form =N-N=O, where the N=O group is bonded directly to a nitrogen) have been shown to be powerful carcinogens in every species of laboratory animal tested. Nitrosamines are alkylating agents which primarily alkylate (add methyl or ethyl groups to) the N7 and O6 atoms of guanine, leading to GC-to-AT point mutations. O6 alkylations are less numerous, but more mutagenic than N7 because O6-methylguanine readily mispairs with thymine.
Sodium nitrite (NaNO2) is used in meats
to prevent growth of Clostridium botulinum bacteria, which produces the
potent botulism neurotoxin. Potentially, sodium nitrate could react with hydrochloric
acid in the stomach to produce nitrous acid, which could in turn react with dimethylamine
to produce carcinogenic dimethylnitrosamine (also known as
N-nitrosodimethylamine). Nitrous acid from nitrosamines, nitrites and nitrates can
deaminate cytosine, adenine and guanine in DNA & RNA.
But levels of dimethylamine or nitrate in the diet may not be sufficient to produce
much dimethylnitrosamine. Only about 10% of dietary nitrate comes from nitrate used
as a preservative. A study in France found no association between dietary intake of
nitrate or nitrite and gastric cancer [EUROPEAN JOURNAL OF EPIDEMIOLOGY 11:67-73
(1995)].
| Dimethylnitrosamine | Nitrous Acid reacts to produce Dimethylnitrosamine |
|---|---|
![[Dimethylnitrosamine]](G_dmns.gif)
|
![[Formation of N-nitrosodimethylamine]](Gnitroso.jpg)
|
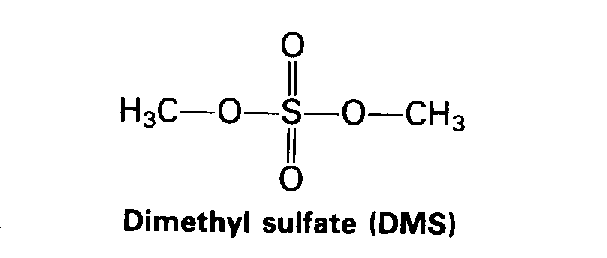
|
Unlike other alkylating agents, DiMethyl Sulfate (DMS) alkylates DNA almost exclusively at nitrogen sites (O6-methylguanine accounts for less than 0.2% of the alkylation). The mutagenicity of DMS is believed to be due to errors in DNA repair. DNA damage due to DMS is normally fixed by Base Excision Repair (BER) enzymes.
As described in the section on epigenetic dysregulation, cytosine is readily methylated at CpG dinucleotides (Cytosine connected to Guanosine through a phosphate), and nitrous acid or nitric oxide readily deaminates 5-methylcytosine to thymine — an extremely common sequence of events in colon carcinomas. Alterations of a single base (point mutations) account for about 80% or p53 tumor suppressor gene mutations. Mutations of p53 are present in more than 90% of small-cell carcinomas and over 50% of non-small-cell carcinomas of the lung. Sunlight readily turns photochemical smog (NO2) into nitric oxide (NO) which can deaminate methylated cytosine to thymine — and tobacco smoke contains nitrosamines which can also deaminate. Any number of irritant chemicals & particles in tobacco smoke can lead to chronic inflammation of lung tissue activating nitric oxide synthetases which enhance the rate of deamination [TOXICOLOGY LETTERS (82/83):1-7 (1995)].
Other chemicals are metabolized to carcinogens which are free radicals. However, free radical activity forms only a small part of chemical carcinogenesis.
TOBACCO 33 %
DIET AND ADULT OBESITY 32 %
PERINATAL EFFECTS 6 %
BIOLOGIC AGENTS (EG VIRUSES) 5 %
--------------------------------
OCCUPATIONAL FACTORS 5 %
ALCOHOL 3 %
SEDENTARY LIVING 3 %
REPRODUCTIVE FACTORS 3 %
--------------------------------
INHERITED GENES 3 %
IONIZING AND UV RADIATION 3 %
ENVIRONMENTAL POLLUTION 2 %
FOOD ADDITIVES (INCL. SALT) 2 %
MEDICAL PRODUCTS/PROCEDURES 1 %
%TOTAL %DEATHS %
ORGAN SYSTEM CANCER PER 5-YEAR
DEATHS NEW CASE SURVIVAL
LUNG 38.4 89 13.4
COLON AND RECTUM 10.5 37.6 61
BREAST 8.6 25.3 83.2
PROSTATE 7.0 19 65.8
------------------------------------------------
PANCREAS 4.8 95.9 3.6
STOMACH AND ESOPHAGEAL 4.5 69.7
GYNECOLOGIC 4.5 32 60
NON-HODGKINS LYMPHOMA 3.9 47.1 51
------------------------------------------------
LEUKEMIA 3.5 66.8 50
LIVER 2.4 82
BRAIN AND CNS 2.3 72
KIDNEY 2.1 40.9 57.9
------------------------------------------------
URINARY BLADDER 2.0 20.7 80.7
MULTIPLE MYELOMA 1.8 77.2
SKIN (INCLUDING MELANOMA) 1.7 1.3 86.6 (melanoma only)
ORAL (MOUTH,LIP,TON,PHAR) 1.5 26.8
(For more recent statistics, see my essay
Causes of Death.)
Based on organ system and tissue, there are over a hundred different types of cancer. The suffix -oma means tumor, and benign tumors are denoted by a tissue-name prefix added to the "-oma" suffix. Thus, a benign fibrous tumor is called a fibroma, a benign bone tumor is called an osteoma, a benign fat tumor is called a lipoma and a benign glandular tumor is called an adenoma. Despite being benign, adenomas can be a health-hazard due to their potential for excessive hormone production. Adenoma of the adrenal cortex, for example, can cause Cushing's Disease, and adenoma of the growth-hormone producing cells of the anterior pituitary can cause acromegaly.
Malignant cancers can be classified into three major groups: (1) carcinomas, malignancy of epithelial cells — which accounts for nine-tenths of all cancers, (2) sarcomas, malignancy of mesodermal cells — usually connective tissue, and (3) leukemias & lymphomas, which are malignant cancers of the white blood cells, blood-forming tissue and lymph tissues.
Lung cancer is the leading cause of death due to cancer for both men and women, accounting for 32% of male cancer deaths and 25% of female cancer deaths in the United States. Only about one tenth of lung cancer victims survive more than 5 years after diagnosis. About one-third of lung cancers are adenocarcinomas, about one-third are squamous-cell carcinomas, about one-fifth are small-cell carcinomas and about one-tenth are large-cell carcinomas.
Small-cell carcinoma has the best chance of being cured by surgery (a nearly 30% 5 year survival rate), and small-cell carcinoma is the most responsive to chemotherapy. About 90% of small-cell carcinomas are associated with p53 mutations and about 90% are associated wit Rb mutations, in contrast to 50% and 20% respectively for non-small-cell carcinomas. Bcl-2 overexpression is seen in about three-quarters of small-cell carcinomas and about half of non-small-cell carcinomas.
Lung cancer, like pancreatic cancer, is one of the most infrequently cured of all cancers. Prevention is the greatest defense against lung cancer death. Currently, at least 90% of all lung cancers (and 40% of all cancers) are due to cigarette smoking. Smoking also accounts for 25% of all heart attack deaths, but a smoker who quits halves the heart-attack rate within one year of quitting. The effect of quitting on lung-cancer rates is a much slower decline. Nonsmoking wives of smokers have twice the incidence of lung cancer as nonsmoking wives of nonsmokers. An estimated 2-3% of lung cancer deaths are attributed to second-hand smoke.
Cigarette smoking was rare at the turn of the century (as was lung cancer), but by 1935 fully 66% of American men and 26% of American women were smokers. Smoking maximized for women at 35% in the mid-1960s, at a time when smoking among men had dropped to 50%. By 1978 27% of women and 32% of men smoked. The decline was greatest for college graduates, 16% of whom were still smoking in 1987. Increased use of cigarettes low in tar and nicotine has caused smokers to smoke more cigarettes and inhale more deeply — increasing rates of lung adenocarcinoma, compared to squamous cell carcinoma [ONCOGENE; Shields,PG; 21(45):6870-6876 (2002)].
The urine of smokers is full of mutagens (which helps explain why smoking increases cancer rates in the urinary bladder and so many other organs). It is easy to induce cancer on the skin of rabbits (but not guinea pigs) by painting them with the tars condensed from cigarette smoke. Interestingly, tobacco smoke has never conclusively been demonstrated to cause lung cancer in experimental animals.
Air pollution is deemed to be responsible for 2-3% of lung cancers. Asbestos also has been a significant causative agent, although this is expected to decline with reduced asbestos use. Asbestos probably acts as a promoter (mitogen), complementing the mutagenic substances (inducers) in tobacco smoke. The combination has multiplicative rather than additive effects. Whereas smoking can increase the chance of lung cancer by 10 and asbestos can increase the chance by 5, the combination can increase the chance by 50.
Estimates of Radon as a cause of lung cancer death are controversial. The US Environmental Protection Agency (EPA) has put the figure as high as 5-20%, but only on the basis of questionable extrapolations based on uranium miners. A typical square mile of topsoil (one foot deep, 1.7 million tons) contains about 6 tons of Thorium and 4-5 tons of Uranium. Radioactive decay causes the colorless, odorless gas Radon to be released into water & air from the soil under homes. Because Radon is chemically inert, it readily passes through many materials. The EPA has estimated that nearly one American home in 15 contains excessive amounts of Radon gas. Ironically, energy conservation measures worsen the problem by reducing ventilation. Running hot water in showers and dishwashing can release more Radon gas. Radon testing devices can be purchased from many hardware stores, and there are professionals who can reduce the Radon content of homes.
The term large bowel is synonymous with "large intestine", being composed of both the colon and the rectum. The colon constitutes approximately 90 cm (3 feet) of the large bowel, whereas the rectum constitutes the last 15 cm (6 inches). Cancer of the large bowel is the second most common cause of cancer death in the United States, after lung cancer. Rectal cancer is approximately one-seventh as common a cause of death as colon cancer, although it accounts for nearly half of the diagnosed new cases.
Most colorectal cancers arise from adenomatous polyps (a polyp is a grossly visible protrusion from the mucosal surface). Adenomatous polyps are present in about a third of adults middle-aged or older, but fewer than 1% of polyps become malignant. A polyp only has a significant chance of becoming malignant if it is 1.5 to 2.5 cm in size (2 to 10% chance) or larger than 2.5 cm (10% chance). Most colorectal cancers follow a five-step process of adematous polyps which includes oncogene mutational activation followed by tumor suppressor gene inactivation: (1) point mutation of K-ras proto-oncogene (2) hypomethylation of DNA, leading to gene activation (3) DNA loss of the APC [Adenomatous Polyposis Coli] tumor-suppressor gene (4) DNA loss of the DCC [Deleted in Colorectal Cancer] tumor-suppressor gene (5) DNA loss of the p53 tumor-suppressor gene. Non-hereditary forms of colorectal cancer are usually characterized by aneuploidy (an abnormal number of chromosomes). Colorectal cancers having a normal number of chromosomes typically have microsatellite instability [CELL; Kinzler,KW; 87(2):159-170 (1996)].
Between 5 to 10% of colon cancers are inherited. Hereditary forms of colorectal cancer include polyposis coli (an APC gene mutation resulting in thousands of adenomatous polyps) and HNPCC (Hereditary NonPolyposis Colon Cancer), associated with errors in DNA replication due to defective repair of DNA mismatches. HNPCC is nearly always associated with microsatellite instability, most often associated with mutations or epimutations of mismatch repair genes hMlh1 [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); 95(12):6870-6875 (1998) or MHsh2 [JOURNAL OF BIOLOGICAL CHEMISTRY; Mazurek,AM; 277(10):8260-8266 (2002)]. Tumor Necrosis Factor beta (TNF-ß) normally provides cells with signals inhibiting cell growth, but defective DNA mismatch repair leads to a defect in the TNF-ß receptor resulting in the unrestrained cell growth of HNPCC. Overexpression of inducible cyclooxygenase enzyme (COX-2, which forms prostaglandins mediating inflammation) is particularly common in colon cancers initiated by APC gene mutation. Nitric oxide increases COX-2 expression. The phytochemical curcumin (which gives curry its yellow color) inhibits inducible nitric oxide synthetase (thereby inhibiting COX-2). Curcumin has been shown to reduce pre-cancerous colon lesions by 45% [CARCINOGENESIS; Rao,CV; 20(4):641-644 (1999)].
APC protein functions by causing destruction of the transcription factor ß-catenin. Defective APC results in ß-catenin accumulation in the cytoplasm, ß-catenin transcription activity in the nucleus, and over-transcription of the Wnt target genes (producing Wnt proliferation-signalling protein) — leading to the excessive proliferation of polyp formation [SCIENCE; Radtke,F; 307:1904-1909 (2005)].
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) significantly increase expression of the PTEN tumor suppressor gene, thereby increasing apoptosis of cancer cells [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS; Chu,EC; 320:875-879 (2004)].
A 70-year-old is a thousand times more likely to develop colon cancer within a year than a 20-year-old. Regular examinations can detect colorectal cancers before symptoms appear, so colorectal exams are recommended for everyone over 40. Digital examinations can often reveal rectal cancer, but colonoscopy or sigmoidoscopy will be needed for the colon. Because adenomatous polyps require more than 5 years of growth before becoming clinically significant, colonoscopy need only be done every 3 years.
An epidemiological study of water chlorination in Wisconsin which looked only at data for white females found a significant association between municipal water chlorination and colon cancer, but no association with any other form of cancer [JNCI (Journal of the National Cancer Institute) 67(6):1191-1198 (1981)].
Diet appears to play a significant role in the development of colorectal cancer. High saturated (animal) fats and low fiber are said to be causative. Animal fat (meat) is particularly a problem when associated with a low intake of fruits & vegetables. Cereal fiber & legumes may be somewhat protective. But fiber and/or phytochemicals from fruits & vegetables can provide as much as a 3-fold protection for those who eat five or more servings daily (in contrast to those who eat one or less). [see JOURNAL OF THE NATIONAL CANCER INSTITUTE 82:650-661 (1990)]. Vitamin D3 induces expression of cystatin D, leading to tumor suppression of colon cancer [THE JOURNAL OF CLINICAL INVESTIGATION; Alvarez-Diaz,S; 119(8):2343-2358 (2009)].
Of those who die of breast cancer, only about one-half of 1% are males — suggestive of an important role played by hormones. Women who do not have functioning ovaries and who do not receive estrogen replacement do not develop breast cancer. Women who experience puberty at age 12 have nearly twice the lifetime risk of those who have puberty at age 16. Women who have menopause at age 42 (ten years earlier than the median 52) have 35% less breast cancer. Women who have a full-term pregnancy by age 18 have nearly a third the risk of breast cancer of women who never experience childbirth.
In 2002 a large-scale study was halted due to mounting evidence that Hormone ReplacementTherapy (HRT) combining estrogen & progestin in women with a uterus (no hysterectomy) increased strokes 41%, heart attacks 29% and breast cancer 26%, while reducing colorectal cancer 37% and hip fractures 33% [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION 288(3):321 (2002)]. Nonetheless, there is no evidence that oral contraceptives increase the risk of breast cancer (and there is substantial evidence that oral contraceptives are protective against ovarian & endometrial cancer).
Women who inherit one mutant allele of the BRCA1 (BReast CAncer, Type 1) or BRCA2 tumor-suppressor gene have an 85 to 90% lifetime chance of developing breast cancer — in contrast to a roughly 10% lifetime risk for women without cancer gene abnormality. BRCA1 & BRCA2 proteins are required for DNA double-strand break repair, particulary DNA repair by homologous recombination [ONCOGENE; Jasin,M; 21(58):8981-8993 (2002)]. BRCA1 helps repair DNA damage, but the specificity of the gene defect in promoting cancer of the breast (rather than other tissues) is due to the ability of BRCA1 protein to limit progesterone-receptor expression (excessive progesterone stimulation leads to excessive cell growth) [SCIENCE; Poole,AJ; 314:1467-1470 (2006)].
Breast cancers that overexpress the epidermal growth factor receptor (ErbB2) or have a mutated p53 tumor-suppressor gene are resistant to chemotherapy have a bad prognosis. Breast cancer staging is often done by a TNM scheme: Tumor(T), Lymph Nodes(N) and Distant Metastasis(M).
Estogen metabolites formed by cytochrome P450 (CYP) enzymes play a role in the initiation and progression of cancer. CYP1B1 enzyme converts 17β−estradiol to the carcinogenic 4−hydroxyestradiol that forms adducts with DNA and undergoes redox cycling to generate reactive oxygen species that can damage DNA, protein and lipids. The small redox protein thioredoxin is a growth factor overexpressed in breast cancer tumor cells and has been shown to increase expression of CYP1B1 [CARCINOGENESIS; Husbeck,B; 23(10):1625-1630 (2002)].
Screening for breast cancer has been very effective. Regular X-ray mammograms past the age of 40 have reduced the death rate by about 25%. A few women have had their breasts surgically removed and replaced with implants simply as a precautionary measure.
Because both breast & colon cancer are rare in Japan, but are not rare among Japanese-Americans, diet has often been pointed-to as making the difference. Fat constitutes 37% of total calories in the United States, in contrast to 11% in Japan. Saturated fat is associated with a somewhat higher incidence of breast cancer, whereas fruits & vegetables are associated with a lower incidence — but the effect and the association are not strong. Even one alcoholic drink per day increases the risk of breast cancer by 7% to 9%, with an additional 7% to 9% increased risk for each additional drink.
(For detailed breast cancer statistics, see Breast Cancer: Statistics on Incidence, Survival and Screening.)
Prostate cancer (even moreso than colon cancer) is primarily a disease of later life. Over 80% of prostate tumors are found in men over 65 years of age. Prostate cancer is the major cause of cancer death for men older than 75. It is surprising that more elderly men don't opt for prophylactic surgical removal. Blacks in North America have twice the death rate from prostate cancer as white men, and higher blood testosterone levels in black men has been the suggested cause. Japanese men have a lower incidence of prostate cancer when living in Japan, but the insidence increases to the level of American whites upon immigration to the United States, suggesting a role for diet or other environmental factors. Heavy drinking accounts for nearly 20% of all prostate cancers.
Testosterone is converted to dihydrotestosterone by the enzyme 5-alpha-reductase. Estradiol levels normally increase in men with age (at least relative to testosterone) and may lead to prostatic growth (benign prostatic hyperplasia, ie, nonmalignant tissue growth). Increased androreceptor protein due to estrogen acts with dihydrotestosterone to increase hyperplagia. 90% of American men have prostatic hyperplagia when autopsied over age 80. Obstructed urinary flow is common.
A University of Southern California study showed the lowest rate of prostate cancer in men with the lowest levels of 5-alpha-reductase. A study of 51,000 men by the Harvard University School of Public Health showed twice the incidence of advanced prostate cancer among men eating high levels of fat (close to 90 grams daily) as compared to those with lower fat intake (close to 50 grams daily). If the fat was derived from red meat, the factor was 2.6.
The most frequently mutated gene in prostate cancer is the tumor-suppressor gene PTEN (60%-80%) [MEDICAL SCIENCE MONITOR; Chu,EC; 10(10):RA235-RA241 (2004)]. Deletion of the cell cycle regulatory tumor-suppressor genes RB or p16 is seen in about half of prostate cancer cases [CANCER RESEARCH; Jarrard,DF; 59(12):2957-2964 (1999)].
Screening for Prostate Specific Antigen is recommended for men over 50 by the American Cancer Society. Before PSA screening became commonplace, two-thirds of diagnosed prostate cancers were already malignant. Now nearly two-thirds of cancers detected in screening can be cured by surgery or radiation. But PSA testing has not yet had an impact on death rates, in part because the disease often progresses so slowly that men often die of other causes. The test is also controversial because it has a high rate of false positives and false negatives. Biopsy is essential for positive diagnosis. Only a third of biopsies in men with elevated PSAs show cancer, whereas a quarter of prostate cancer patients have normal PSA levels.
Pancreatic cancer has the lowest survival rate of any of the major causes of cancer death. There are usually no symptoms until the disease is very advanced. The median survival time for patients not eligible for surgery is about 6 months. Even those who are treated surgically have a 5 year survival rate of only ten percent.
Over 90% of pancreatic cancers are ductal adenocarcinomas, with the remainder being islet cell tumors. About 70% of pancreatic cancers occur in the head of the pancreas, which often leads to obstruction of the bile duct followed by jaundice, darkening of the urine and a claylike stool. Over 85% of pancreatic cancers contain mutations in the K-ras member of the ras family of oncogenes which cause the release of mutagenic growth signals.
Smoking is the most commonly cited risk factor, with heavy smokers being two or three times more likely to contract the disease than nonsmokers. Incidence is higher in countries with high-fat diets, and fruits & vegetables have been shown to be strongly protective.
In 1930 stomach cancer was by far the leading cause of cancer death among men in the United States, but the incidence of the disease has dropped significantly since then. Canada has one of the lowest stomach cancer rates among all the developed countries. This is in contrast to Japan, where the incidence is 6 times higher. Stomach cancer is the most common cause of cancer death in Japan — a fact often associated with the frequent consumption of pickled, burned and highly-salted foods in that country. Bologna, ham, hotdogs and sausage that are preserved with nitrates often result in highly carcinogenic nitrosamines being produced in the stomach. Stomach cancer is more frequent in patients suffering from ulcers associated with the bacterium Heliobacter pylorii. People with blood type A are roughly 20% more likely to develop stomach cancer than people of type O or type B. About 85% of stomach cancers are adenocarcinomas. Fruits & vegetables have been shown to be strongly protective against stomach cancer.
Smoking & alcohol are believed to be the highest risk factors for esophageal cancer. Although esphageal cancer is rare, it is quite fatal, with fewer than 5% of victims being alive 5 years after initial diagnosis. Fruit is particularly protective against esophageal cancer.
The most common gynecologic cancer in the US is ovarian cancer, which accounts for
5% of all cancer deaths of women in the United States. Each pregnancy reduces the risk
of ovarian cancer by about 10 and oral contraceptives also reduce the risk. Both
pregnancy & contraceptives reduce the number of ovulations, and it is hypothesized that
ovarian cancer arises due to aberrations associated with ovarian epithelial repair after
each ovulation. Women who inherit one mutant allele of the BRCA1 tumor-suppressor
gene have a 55% lifetime chance of developing ovarian cancer. Women who inherit one
mutant allele of the BRCA2 tumor-suppressor gene have a 25% lifetime chance
of developing ovarian cancer. Women without abnormality of BRCA1 or BRCA2 have a
1.8% change of getting ovarian cancer. Ovarian cancer is the
hardest to screen-for, and it is therefore most often fatal.
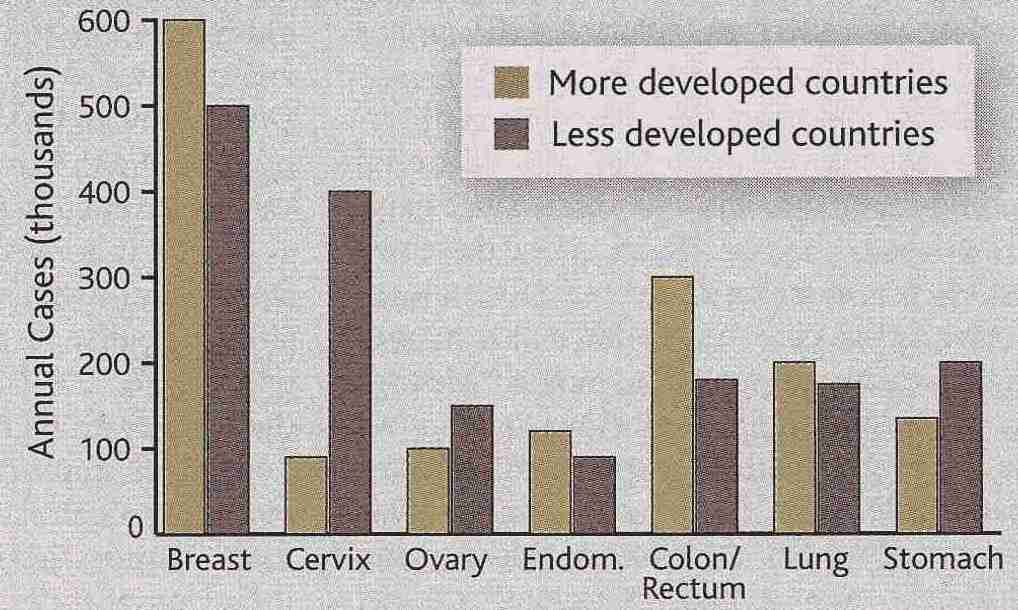
|
The endometrium is the glandular mucous membrane that lines the uterus, so endometrial cancer is the same as uterine cancer. The most frequently mutated gene in endometrial cancer is the tumor-suppressor gene PTEN. Only 25% of cases of uterine cancer occur before the age of 50.
Cervical cancer is among the few human cancers caused by a virus — the Human PapillomaVirus (HPV), some forms of which cause venereal warts. 70-80% of cancers of the genitals & anus are associated with HPV. Unlike other viruses, which thrive in blood or internal organs, HPV prefers skin (epidermal) cells. The HPVs causing cervical cancer make E6 and E7 proteins which, respectively, bind-to (and thereby facilitate the degradation-of) the two tumor-suppressor proteins: p53 and pRb. Inactivation of p53 by E6 is oncogenic because apoptosis by p53 is normally a mechanism for eliminating cancerous cells [WORLD JOURNAL OF GASTROENTEROLOGY; 11(7):931-937 (2005)]. Vaccines against HPV lock onto a protein forming most of the HPV coat. There are many HPV strains, but strains of type HPV−16, HPV−18 and HPV−31 are responsible for 95% of cases of cervical cancer. CpG methylation occurs much more often in the LCR region (non-coding region with many transcripion binding sites) of the HPV genome in HPV−16 & HPV−18 [THE AMERICAN JOURNAL OF FORENSIC MEDICINE AND PATHOLOGY; Takeichi,S; 5(3):223-227 (1984)]
The Papanicolanou smear (Pap smear), which takes a sample from the fluid in the uterus, is 90 to 95% accurate in detecting cervical cancer in the early stages, but is less accurate for advanced malignancies. 85% of those who die from cervical cancer in the US never had a Pap smear in their life. Inflammation & hemorrhage can cause false positives. Pelvic examinations are useful for detection of uterine cancer. The effectiveness and widespread use of Pap smears in developed countries accounts for the great disparity in cervical cancer prevalence between developed and undeveloped countries.
Most studies have shown a significant protective effect of fruits & vegetables for gynecologic cancer.
Hodgkin's Lymphoma is a malignancy characterized by chronic inflammation of lymph nodes, which is usually curable because it is usually localized. Non-Hodgkin's Lymphoma, on the other hand, is usually fatal, with the majority of victims being children or young adults. Only 10% of non-Hodgkin's lymphomas are derived from T-cells, the other 90% coming from B-cells.
Lowering of immune system function is the most common risk factor for Non-Hodgkin's Lymphoma. Immunosuppressive drugs and the rise of HIV infections have caused most of the increased incidence of this disease. Up to 30% of HIV-1 infected persons develop non-Hodgkin's lymphoma, nearly half of those due to the Epstein-Barr virus which causes mononucleosis.
Leukemia, cancer of the leucocytes (white blood cells), is most often myeloid (bone marrow) leukemia affecting the granulocytes, and less often lymphoid (eg, lymph nodes). Chronic Myeloid Leukemia (CML, myeloid = myelogenous = myeloblastic) is characterized by gradual onset of anemia associated with marked enlargement of the spleen, liver or lymph nodes over a period which exceeds one year. By contrast, Acute Myeloid Leukemia (AML, which occurs nearly twice as often) leads to death within months, associated with symptoms of acute infection, severe anemia and hemorrhaging.
Nearly 90% of CML patients have shortened chromosome 22 (the "Philadelphia chromosome", discovered in Philadelphia in 1960), due to translocation to chromosome 9. Leukemia is primarily a defect in cell differentiation rather than cell proliferation. The leukemic cell population increases because the rate of proliferation exceeds the rate of cell elimination. Myeloblast proliferation in AML is often slower than in CML.
About 10% of AML cases are Acute Promyelocytic Leukemia (APL) in which promyelocytic leukemic cells accumulate in the bone marrow and the PML tumor suppressor protein is inactivated, with delocalization of the PML nuclear bodies.
Although most cases of leukemia occur in people over 60 years of age, the disease is most shocking in children, because 85% of such cases are acute — and result in death within a few months. Long term survival of Acute Lymphocytic Leukemia (ALL) is in excess of 70%, but for AML & CML survival is not over 25%.
Leukemia is associated with ionizing radiation more frequently than any cancer. Diagnostic X-rays may cause one-sixth of all cases of leukemia. Japanese survivors of atomic bomb explosions in World War II developed an increase of myeloid leukemias with peak incidence about 6 years after exposure. Nontheless, radiation cancer therapy is associated with increased acute leukemias less often than are chemotherapeutic agents. In fact, leukemia is the most frequent type of cancer induced by treatment with alkylating agents [JOURNAL NUTRITION; Boyonoski,AC; 132(1):108-114 (2002)]. AML is directly correlated with radiation and alkylating agents, whereas the cause of CML is unclear.
Although a few epidemiological studies have resulted in an association of childhood leukemia with electric and magnetic fields (EMFs), others have found negative results. If such an effect exists, it may be too small to unambiguously measure — including the claimed association between EMFs and melatonin levels. [see SCIENCE 358: 1724-1725 (11 Dec 92) and JAMA 268 (5):625-628 (5 Aug 92)].
Liver inflammation (a mitogenic effect), ie hepatitis, is the most commonly associated risk factor for liver cancer. Hepatitis B is a strongly proven carcinogen. In fact, the widespread prevalence of Hepatitis B and C in developing countries make liver cancer the leading cause of cancer worldwide. Aflatoxin, a mold associated with peanuts, is one of the most carcinogenic substances known — and it is regarded as a frequent cause of liver cancer in Africa.
Because neurons are non-dividing cells, brain cancer is only associated with glial cells (which are dividing cells). Peak incidence of the disease is in childhood or between the ages of 50 & 70, affecting males at least twice as frequently as females. There are no specific risk factors.
One third of kidney cancer cases are deemed to be caused by smoking, probably due to the large number of mutagens found in the urine of smokers. Obesity is also a risk factor. Kidney cancer strikes men twice as often as it strikes women.
Urinary bladder cancer is primarily caused by agents in the urine that are in contact with the bladder wall — including carcinogens entering the body in cigarette smoke. Cigarette smokers have 2-3 times the incidence of bladder cancer as non-smokers. For men, 50% of the cancers are due to smoking and 25% are due to occupational exposures to carcinogens. Polycyclic aromatic hydrocarbons are the primary chemicals increasing risk. Fruit & vegetables have been shown to be highly protective.
Multiple myeloma is cancer of the plasma cells (myeloma cells) that remain in the bond marrow and produce antibodies. There are no known risk factors for this disease.
Skin cancer is so common, and so rarely fatal, that it often is excluded from cancer statistics. The exception to this is melanoma, which is cancer of the melanin (pigment) producing cells (melanocytes) of the epidermis — often concentrated in areas that produce freckles or moles. Although melanoma represents only 5% of diagnosed skin cancers, it causes 75% of skin cancer deaths. Melanoma that has metastasized is generally incurable. The brain is a common site for melanoma metastasis. Whites develop melanoma 40 times more frequently than blacks. Dark skin is generally protective. The most vulnerable are those with fair complexion & freckles who sunburn easily.
Most skin cancers (including melanoma) are caused by sunlight, primarily Ultraviolet-B (UV-B, 290-320nm). Although basal cell carcinomas account for nearly 80% of nonmelanoma skin cancers, squamous cell carcinomas account for most of the deaths because of a greater ability to metastasize.
Childhood sun exposure is the major risk factor for skin cancer, which is permanent and cumulative. It has been estimated that 80% of a person's lifetime sun exposure occurs before the age of 18. Skin cancer was rare at the turn of the century, but so was sun-bathing. When people did go swimming, they took protective clothing (much the way Arabs still protect themselves). Even hat-wearing became unpopular in the 1960s. At the same time the ozone layer of the atmosphere has thinned. Sunlight is always damaging to the skin, even though it is a major source of vitamin D3. The less sun exposure, the less the risk of skin cancer and the less skin-wrinkling.
A change in the size, color or texture of a mole is cause for examination by a physician.
A 10-cigarette-per-day smoker who consumes more than 7 ounces of alcohol daily has a 5-fold increase in the risk of these cancers. More than a pack of cigarettes smoked per day results in a 24-fold increase. The effects of alcohol & smoking are multiplicative, rather than additive. Bad teeth & chronic dental irritation have also been associated with oral cancer. Epstein-Barr virus has frequently been associated with nasopharyngeal cancer.
Most patients have lymph node involvement or distant metastases. The prognosis is generally poor. Hoarseness is often a symptom of laryngeal cancer and the failure to respond to antibiotics should be a warning.
Fruits have been shown to be particularly protective in these cancers.
Specific comments have been made concerning risk factors under the specific organ-associated cancers described in Section VI above. Diet, physical activity, sun-exposure and consumption of tobacco & alcohol are the most general controllable risk factors for cancer (occupational exposures, and environmental factors at certain locations are more specific). Reduction of tobacco & alcohol consumption and increase in physical activity are recommended to reduce cancer risk. Cessation of tobacco use is the single most decisive step any smoker can take to reduce risk of cancer. A smoker has twice the likelihood of dying in any given year as a nonsmoker after the age of 40 — and only 26% of smokers live to age 80 — in contrast with 57% of nonsmokers [ADDICTION 97:15-28 (2002)]. Cessation of alcohol use can also reduce cancer risk significantly. Each alcoholic drink per day increases breast cancer risk by an additional 9%, and risk for other cancers increases as well.
Although carcinogenic (cancer-causing) chemicals can enter the body through inhalation & drinking, diet is a route of entry that affects everyone. The main dietary carcinogens are nitrosamines, polycyclic aromatic hydrocarbons and heterocyclic amines. Meat is particularly carcinogenic when it is cooked in such a way that protein & fat-containing juices drip from the meat onto a hot surface, which generates fumes that are re-deposited upon food. Meat juices used in sauces make the sauces more carcinogenic. Fried eggs can similarly be quite carcinogenic/mutagenic.
Since the 1980s, there has been a great increase in consumption of low-fat foods under the assumption that they are less fattening. Low-fat foods are often very high in sugar/carbohydrate and are, in fact, being blamed for the great increase in obesity since the 1980s. Increased obesity & increased insulin-resistance due to high sugar/carbohydrate diets are believed to increase risk of cancer, particularly colon cancer. An epidemiological study of nearly one million adults followed for sixteen years showed a very strong correlation between obesity & cancer, but the mechanisms remain elusive and it has not been proven that weight-loss is a means of reducing the risk of cancer [NEW ENGLAND JOURNAL OF MEDICINE 348(17):1623-1638 (2003) and 349(5):502-504 (2003)].
Another dietary trend, this one for many decades, has been the increased consumption of omega-6 fatty acids — in contrast to omega-3 fatty acids and saturated fat, especially in vegetable oils used in margarine and other foods, such as safflower & sunflower oil. The resultant increases in chronic inflammations & allergies has contributed to pancreatitis/pancreas-cancer, colitis/colon-cancer, esophagitis/esophageal-cancer, etc. A diet higher in omega-3 fatty acids and lower in the omega-6s could do much to reduce these inflammation-related cancers. Gamma-tocopherol (the major form of Vitamin E in food, in contrast to alpha-tocopherol, which is the major form of Vitamin E in supplement pills) blocks cyclo-oxygenase and reduces proinflammatory PGE2 & LTB4 formation [FASEB JOURNAL 17:816-822 (2003)].
Many studies looking specifically at colon or colorectal cancer find a high cancer risk with red meat (beef, pork, lamb), but no such association with white meat (poultry) or fish [AMERICAN JOURNAL OF CLINICAL NUTRITION 66(suppl):1564S-1571S (1997)]. Diets high in red meats show a 3-fold increase in fecal N-nitroso compounds, whereas fish & white meat show no such increase. Milk, butter and cheese consumption is rarely associated with colon cancer, despite the fact that they contain the same profile of saturated fatty acids as beef.
A high-fiber diet is associated with reduced incidence of both hemorrhoids and colorectal cancer. It has been estimated that an average 70% increase in food fiber would reduce colorectal cancer by nearly one-third in the United States [JOURNAL OF THE NATIONAL CANCER INSTITUTE 84(24):1887-1896 (1992)].
Diets high in fruits & vegetables are often very protective against cancer. Foods rich in Vitamin C, Vitamin E and polyphenols have been shown to inhibit the production of carcinogenic N-nitroso compounds in humans. The results of case-controlled and cohort studies of the protective role of fruits & vegetables against various cancers are presented in the following table from [AMERICAN JOURNAL OF CLINICAL NUTRITION 78(suppl):559S-569S (2003)].
| CANCER | CASE-CONTROLLED | COHORT | |
|---|---|---|---|
| Espophagus | Fruit & Vegetables | ||
| Lung | Fruit & Vegetables | Fruit | |
| Stomach | Fruit & Vegetables | ||
| Colorectum | Fruit & Vegetables | ||
| Breast | Vegetables | ||
| Bladder | Fruit | Fruit |
Cohort studies are regarded as being more reliable than case-controlled studies because the latter are more subject to reporting or recall which is biased by knowledge of the cancer. Nonetheless, significant risk reduction seen in case-controlled studies should not be dismissed, nor should non-significant risk reduction seen in cohort studies.
Cruciferous vegetables such as broccoli, cauliflower and Brussel sprouts contain isothiocyanates (such as sulforaphane) which have been shown to reduce cancer risk in laboratory animals. Sulforaphane is believed to act by inducing Phase 2 enzymes which detoxify many carcinogens along with Reactive Oxygen Species (ROS). However, sulforaphane can also induce Phase 1 enzymes, which can activate carcinogenic polycyclic aromatic hydrocarbons. Thus, smokers and others exposed to high levels of environmental carcinogens may actually worsen their cancer risk by consumption of cruciferous vegetables [CARCINOGENESIS 25(1):61-67 (2004)]. Sulforaphane is also a potent inhibitor of the bacterium Helicobacter pylori, which is a principle cause of gastric cancer — the leading cause of cancer death in Japan and most of the developing world [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 99(11):7610-7615 (2002)].
Bruce Ames (originator of the Ames test, which uses bacteria to screen for mutagenicity of chemicals) advocates the use of supplement vitamin/mineral pills as "insurance" against insufficient micronutrients from insufficient fruit & vegetable consumption. Although the RDA (60mg/day) of Vitamin C is sufficient to prevent scurvy, Ames estimates that 100mg/day is necessary to minimize DNA damage. Deficiency of folate and Vitamin B12 can result in deficient methylation of uracil to thymine and subsequent incorporation of uracil into DNA resulting in chromosome breaks [TOXICOLOGY LETTERS (102/103):5-18 (1998)].
There has been a great deal of controversy over the effectiveness of animal testing to establish whether a substance causes cancer. In 1977 the FDA banned saccharin on the basis of tests that showed an increased incidence of bladder cancer in male rats with dosages equivalent to a human being drinking 1,000 cans of diet soda (55 pounds of sodium saccharin) daily. Since then, a great deal of evidence has established that saccharin is not a carcinogen for humans.
Many substances can be harmful in both insufficient and excessive quantities, including salt, iron, selenium, cholesterol and vitamin A. The assumption of a linear dose-response effect is particularly questionable for mitogens — a low dose may produce no mitogenesis at all. On the other hand, X-rays probably produce tiny amounts of damage even with the tiniest dosages.
The eminent scientist Bruce Ames helped reduce the necessity of giving large doses of substances to laboratory animals as a means of studying carcinogenicity. He developed a test (the Ames Test) using Salmonella typhimurium bacteria and liver extracts that demonstrates mutatgenicity. Nonetheless, even if all mutagens are carcinogens, the converse is not true. (In recent years, Ames has become something of an anti-Racheal Carson with his claims that fruit & vegetables contain so many more carcinogens & anti-carcinogens than human pollutants that the effects of human pollutants are relatively negligible. Ames does, nonetheless, strongly recommends eating lots of fruits & vegetables.)
Since the 1950s the routine use of pap smears for testing has reduced deaths from cervical cancer 70%. No other diagnostic test for cancer can claim such success. Scanning for colon cancer has been proven to reduce death rate, but the same cannot be said for tests screening for prostate cancer, breast cancer or lung cancer.
Most people's bodies are replete with miniscule, harmless tumors. And the immune system eliminates incipient malignancies on a daily basis. Testing with full-body CT (Computed Tomography) scans would almost certainly find signs of cancer — but the resulting biopsies & panic would usually be counter-productive.
The foremost prostate cancer test measures the blood enzyme Prostate-Specific Antigen (PSA), which rises when the prostate is enlarged. But prostate cancer grows so slowly that the test is often considered useless for men in their 70s — who will probably die before the disease progresses very far. The test is of greatest value for men in their 50s.
Although yearly mammograms have been advised for women over 40, there is no evidence that the test saves lives. (Ductal lavage, however, yields much more definitive results for breast cancer.) Because cancer incidence accelerates with age, cancer testing for most cancers before the age of 50 is usually not recommended except in cases of high risk.
X-ray tests for lung cancer are of little use — by the time tumors are large enough to be observed, they are too large to be treated. Spiral CT scans detect lung cancer malignancies earlier, but result in a very large number of false positives — and there is no proof that early detection extends survival.
In the 1930s fewer than 20% of those diagnosed with cancer had a 5-year survival, but by 1993 this figure had risen to 40%. Still, it is common to hear that billions of dollars spent on cancer research have done little to increase the cancer cure-rate. Improved screening & earlier diagnosis may indeed have done more to improve the treatment success rate than fundamental understanding of the disease. Dramatic declines in death rates for breast, prostate, colon & rectum cancer in recent years are primarily due to more advanced diagnostics and earlier detection. But I believe that this picture will change radically in the next 5-10 years.
Surgery is still the most effective cancer treatment, although radiation and chemotherapy are becoming increasingly more effective. Radiation treatment is likely to become even more effective as the techniques for imaging & targeting cancer cells improve.
Traditional chemotherapeutic agents primarily act against all cells, particularly dividing cells. Since cancer cells tend to be dividing at much greater rates than normal body cells, they are more damaged by these agents. Methotrexate, for example, resembles folic acid enough to bind to the enzyme that converts folic acid into the DNA precursors Adenine and Guanine, thereby inhibiting construction of DNA. Alkylating agents, such as chlorambucil, however, damage all DNA. Cancer patients treated with alkylating agents have a risk of developing secondary leukemia which is between 10 and 100 times greater than the general population (which can be greatly worsened by a reduction in DNA repair capability caused by niacin deficiency [JOURNAL NUTRITION; Boyonoski,AC; 132(1):108-114 (2002)].
The most successful chemotherapy agent is Taxol, which targets microtubules. Taxol has been used with good results for breast, ovarian and lung cancers — although it is not effective for most colon cancers. Side effects for chemotherapy can be severe insofar as it suppresses the immune system and can cause a breach in the blood-brain barrier — in many cases the "cure" is worse than the disease.
Most current cancer drugs were found by randomly screening thousands of chemicals for anti-cancer effects. But Gleevec was discovered by deliberate design. Chronic Myelogenous Leukemia (CML) is caused by abnormal fusion of the two oncogenes BCR & ABL. The kinase (enzyme that transfers a phosphate from ATP, ADP, etc.) produced by these fused oncogenes — the Abl protein — is abnormally active. Mutant Abl causes white blood cells to divide incessantly, but Gleevec occupies the site on the Abl molecule which normally binds to ATP, thereby selectively killing CML cells. Unfortunately, the cancer too often further mutates the Abl protein to prevent Gleevec binding.
PS-431 is an experimental drug that blocks the proteasome, a barrel-shaped enzyme that dismembers damaged or short-lived proteins in cells (a cellular "garborator"). Cancer cells are more vulnerable to the build-up of abnormal proteins than normal cells are.
The oncogene that produces epidermal growth factor (EGF) can produce excess EGF in cancer. EGF is found in large amounts in more than 50% of solid tumors. Many cancers are also associated with large amounts of EGF receptors. The anti-cancer drug Erbitux by ImClone blocks EGF receptors. Despite the fact that thousands of desperate cancer patients had asked for compassionate pre-approval use of Erbitux, the FDA refused to look at ImClone's application and demanded expensive "effectiveness" studies. There is a difficulty in determining which patients are most responsive to anti-EGF drugs.
Other drugs under development — including thalidomide — target the growth factor for blood vessel growth. thalidomide also inhibits the cytokine Tumor Necrosis Factor (TNF) and like Non-Steroidal, Anti-Inflammatory Drugs (NSAIDs) thalidomide can be used to oppose the inflammatory processes believed to contribute to cancer progression [THE LANCET 357:539-545 (2001)]. Agents that inhibit the pro-inflammatory inducible transcription factor NF-kappaB may also prove to be of value against inflammation-aggrevated cancer [ THE JOURNAL OF CLINICAL INVESTIGATION; 107(2):135-142 (2001)]
Platinum-based drugs bond covalently to purine DNA bases, leading to apoptosis of tumor cells. When first introduced, the platinum-based drug cisplatin changed the cure rate for metastatic testicular cancer from 5% to 60%. Testicular cancer is particularly vulnerable to platinum-based drugs because testicular tumor cells have a low Nucleotide-Excision Repair (NER) type of DNA-repair capability [NATURE REVIEWS:CANCER ; Kelland,L; 7(8):573-584 (2007)].
The immune system is primarily effective against neoplasms due to viral infections (a very small proportion of human cancers in developed countries). Otherwise, the immune response does little more than weed-out those mutated cancer cells that fail to masquerade as native tissue. A vaccine developed by Cell Genesys uses the patient's tumor cells to stimulate the immune system. Interferon, a protein produced by cells in response to virus, increases activity of the immune system's killer cells. Patients treated with interferon alpha-2b after surgical removal of tumors have shown a reduced risk of relapse & death in the range of 50%.
Radioimmunotherapy has proven to be both safe and highly effective against Non-Hodgkin's Lymphoma. Zevalin and Bexxar are new drugs that combine genetically engineered monoclonal antibodies (antibodies produced from cells that can be grown indefinitely, derived from a single clone of plasma cells) with radioactive isotopes (Yttrium-90 or Iodine-131). The antibodies bind to the CD-20 antigen found on the surface of B-cells, which proliferate in Non-Hodgkin's Lymphoma. Once attached, the isotopes kill the cell.
Significant advances are also being made in delivery of anti-cancer drugs in such a way as to specifically target cancer cells. Chemotherapy drugs modified to stick to tiny magnified particles can be pulled to the cancer site with a magnetic field. Radioactive isotopes of iodine and yttrium have been coupled with antibodies that stick specifically to cancer cells. Light-sensitive drugs (usually porphyrins) can be drawn into cancer cells and activated with laser light of specific frequency (possibly delivered by a fiber-optic endoscope). Vaccines can inactivate HPV (Human Papilloma Virus).
Gene therapy and other molecular methods are likely to make the big difference in cancer treatment in the coming years. Already an artificial RNA virus has been developed which replicates in cancer cells with an activated Ras signaling pathway (about 30% of all human cancers), but does not survive in normal cells. Similar viruses that carry p53 and other tumor-suppressor genes into cancer cells would do no harm to normal tissues. Some oncolytic viruses are engineered to be "armed" to induce cancer antigens in T−cells, anti-angiogenic factors or anti-cancer cytokines [BMJ; Chernajovsky,Y; 332:170-172 (2006)].
Genes that restore DNA-repair would likewise do no harm to normal cells — and this technique could be parlayed into an anti-aging method. Targeting of the telomerase enzyme also has the potential to combat cancer, but may lead scientists to learn how to "immortalize" normal cells. Anti-angiogenic drugs that stop cancer growth by disrupting the blood supplies to solid tumors are showing great promise, although they are less effective against tumors in which the p53 tumor suppressor gene has been inactivated because p53 loss makes cells more resistant to hypoxia. (p53 is inactivated in bout 50% of cancers.)
Cancer cells often rely on glycolysis rather than oxidative phosphorylation for ATP generation (the Warburg effect). Whether this is due to the necessity of rapid growth in an anaerobic environment, as a strategy for circumventing apoptosis or for other metabolic reasons has not been determined, although glycolysis is known to reduce tumor sensitivity to many anticancer agents. Inhibition of glycolysis with 3−bromopyruvate leads to cancer cell death by both necrosis (energy depletion) and apoptosis [CANCER RESEARCH; Xu,R; 65(2):613-621 (2005)]. Non-cancer cells in many tissues can rely on alternate energy sources that bypass glycolysis (notably fatty acids and amino acids), but this is not possible for tissues heavily reliant on glucose, such as brain, retinae and testis [ONCOGENE; Pelicano,H; 25:4633-4646 (2006)].
Molecular diagnosis of cancer that looks
for oncogenes & tumor-suppressor genes in body fluids will greatly
improve screening in the near future. And there is cause for the optimistic
thought that much other knowledge discovered in the molecular war against
cancer will advance molecular anti-aging techniques. Both effects will
be good news for those who want to live a long, long time.
![[GO TO BEN BEST'S HOME PAGE]](../homeback.gif) HOME PAGE
HOME PAGE