The practice of cryonics is based on the belief that future medicine will be able to restore well-preserved legally dead persons (cryonics patients) to life and youth. Specifically, it is believed that future medicine will be able to cure all disease and repair damage due to aging, the dying process, the cryopreservation process, and some amount of ischemia/reperfusion injury that occurs after legal death [REJUVENATION RESEARCH; Best,BP; 11(2):493-503 (2008)].
How much ischemic damage short of complete decomposition allows for the possibility of future restoration of a person with memories and personal identity intact is the subject of considerable questioning among those who believe that cryonics may work. Attention is focused on the brain, specifically the blood vessels, neurons, and connections between neurons. How well are these components preserved under the typical post-mortem conditions of a cryonics patient.
Most cryonics patients roughly fall into one of the following categories
The terminal patient is pronounced dead immediately after cardiac arrest, and a team waiting by the bedside immediately begins cooling and restoration of circulation. After cooling to at least 10ºC the patient is perfused with cryoprotectants to prevent ice formation in the brain, and is then cooled to liquid nitrogen temperature for long-term storage. Patients with local standby generally suffer mininal post-mortem ischemic damage.
Cooling is begun soon after cardiac arrest, blood is washed-out, and hte patient is shipped in ice to the cryonics facility, experiencing 12 to 36 hours of cold ischemia before cryoprotectant perfusion.
Variable amounts of warm ischemia are experienced prior to cooling, and cooling is often inefficient once begun — without restoration of circulation. A remote patient will spend 12 to 72 hours in ice pending and during shipment to a cryonics facility before cryoprotectant perfusion.
Variable amounts of warm ischemia and cold ischemia are experienced before the patient is placed in dry ice. Once at the cryonics facility the patient is cooled to liquid nitrogen temperature without cryoprotectant perfusion. Although dry ice patients experience severe freezing damage, some cryonicists believe that future nanomedicine will be able to repair the damage or conduct sufficient nanoneuroarcheology to be able to restore the original person with memories and identity intact [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Drexler,KE; 78(9):5275-5278 (1981)].
Few cryonics patient receive the ideal treatment of local standby. It is of great interest to practicing cryonicists to find means to minimize ischemic damage in non-ideal cryonics cases, and to understand the prospects for future reconstruction of patients who have suffered warm and cold ischemic damage. Emphasis is on ischemic damage to brain blood vessels and neural tissue. Undamaged blood vessels may not be of importance for future reconstruction of memory and identity, but it is important to understand if cryoprotectant perfusion can cause reperfusion injury to nervous tissue, or if ischemically damaged blood vessels prevent cryoprotectants from reaching and saturating brain tissue during cryoprotectant perfusion. Ischemically-damaged cryonics patients frequently exhibit edema during cryoprotectant perfusion. Edema impedes perfusion, preventing cryoprotectants from reaching and saturating brain tissue.
If circulation is restored in less than a few minutes
after cardiac arrest, blood vessels and tissues benefit
from the restoration gas exchange and nutrient,
and are able to recover. But if circulation is restored
after a certain delay, blood vessels and tissues are
damaged even more than they would have been without
restoration of circulation. Instead of oxygen
reviving the tissues, oxygen in delayed reperfusion
causes free radical damage.
| Reperfusion injury | Anti-oxidant action | Free radical burst |
|---|---|---|
|
|
|
|
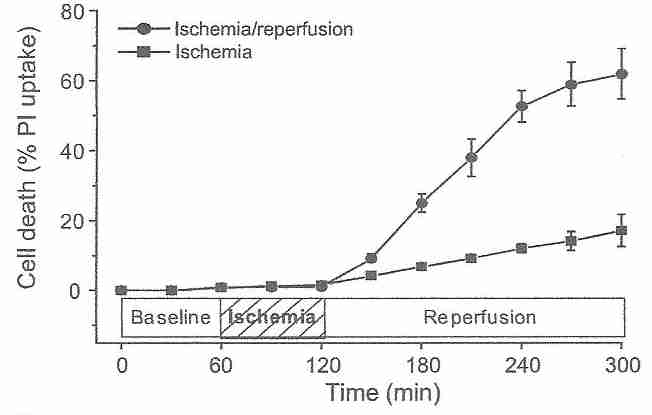
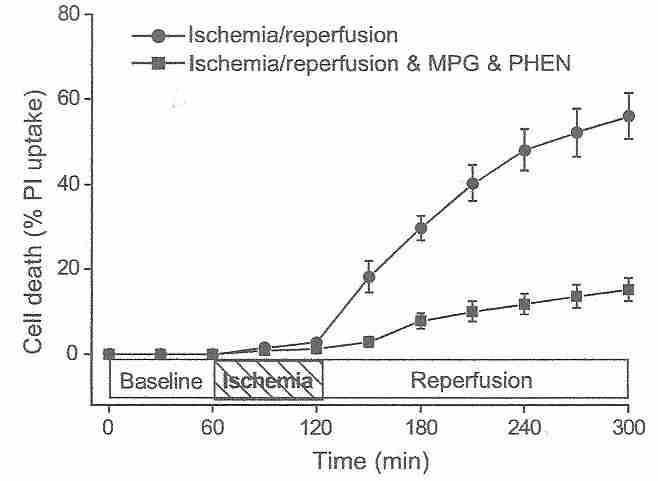
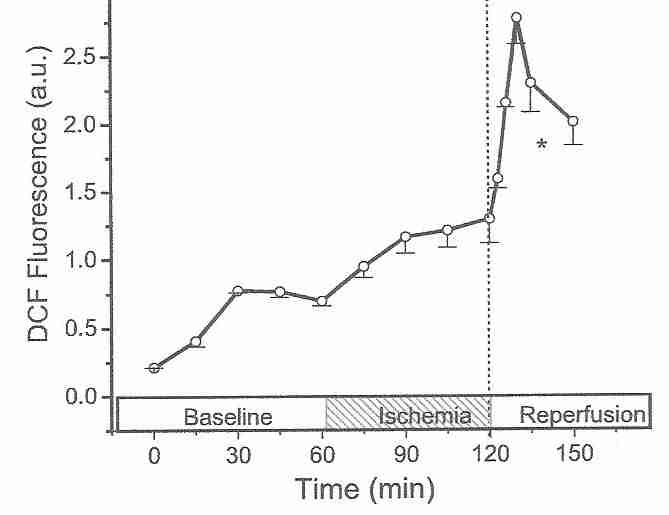
Cardiocytes are much more resistant to ischemic damage than neurons [ JOURNAL OF CLINICAL INVESTIGATION; Lee,J; 106(6):723 (2000)], but the principle of reperfusion damage is the same for both cell types. After an hour of ischemia, 3 hours of reperfusion kills over 60% of cardiocytes, whereas after an additional 3 hours of ischemia (rather than reperfusion), only about 15% of the cardiocytes die [AMERICAN JOURNAL OF PHYSIOLOGY;Vanden Hoek,TL; 270(4 Pt 2):H1334-H1341 (1996)]. The reperfusion injury is completely abolished by antioxidant treatments, demonstrating that reperfusion injury is initially due to oxidative damage [JOURNAL OF MOLECULAR AND CELLULAR CARDIOLOGY; Vanden Hock,TL; 29(9):2441-2450 (1997)]. Like antioxidants, spin-trapping agents protect against the initial stages of reperfusion injury [ JOURNAL OF CEREBRAL BLOOD FLOW & METABOLISM; Aronowski,J; 17(10):1048-1056 (1997)]. The fluorescent oxidant 2',7'-dichlorofluorescin diacetate (DCF) probe for hydrogen peroxide and superoxide shows a rise in ROS (Reactive Oxygen Species) under ischemia, but a dramatic burst of ROS upon initiation of reperfusion [JOURNAL OF MOLECULAR AND CELLULAR CARDIOLOGY; Vanden Hock,TL; 29(9):2571-2583 (1997)]. Similar experiments on rabbit hearts demonstrate a peak in ROS at 10-20 seconds of reperfusion following 10 to 30 minutes of ischemia, with no ROS observable after 5 or more minutes of reflow [JOURNAL OF BIOLOGICAL CHEMISTRY; Zweier,JL; 264(32):18890-18895 (1989)].
Many of the oxidants released on reperfusion were generated during the ischemic period. Reducing the length of ischemia can reduce the level of oxidants produced during reperfusion [JOURNAL OF MOLECULAR AND CELLULAR CARDIOLOGY; Vanden Hock,TL; 29(9):2571-2583 (1997)]. Endothelial cells are rich in mitochondria, and it is mitochondria which are the source of most of the superoxide produced in reperfusion [BIOCHEMICA ET BIOPHYSICA ACTA; Liu,B; 1794(3):476-485 (2009)]. Endothelial cell NADPH oxidase [JOURNAL OF BIOLOGICAL CHEMISTRY; Li,J; 277(22):19952-19960 (2002)] and xanthine oxidase [ JOURNAL OF CELLULAR PHYSIOLOGY; Terada,LS; 148(2):191-196 (1991)] are also significant sources of superoxide during reperfusion. Superoxide reacts with nitric oxide resulting in peroxynitrite, which is more damaging than the superoxide [NATURE REVIEWS DRUG DISCOVERY; Szabo,C; 6(8):662-680 (2007)]. Methylene blue can protect against reperfusion injury by reducing nitric oxide (and hence peroxynitrite) formation [ CRITICAL CARE MEDICINE; Miclescu,A; 38(11):2199-2206 (2010)].
After free radicals, the second wave of blood-vessel damaging agents in ischemia and reperfusion injury are inflammatory cytokines. Tumor Necrosis Factor-alpha (TNF−α) and InterLeukin-one-beta (IL−1β) appear within hours of ischemia, followed by neutrophils within 4 to 6 hours of ischemia [PROGRESS IN CARDIOVASCULAR DISEASES; Rosenberg,GA; 42(3):209-216 (1999)]. Inflammatory cytokines promote further free radical production while activating PolyMorphoNuclear leukocytes (PMN leukocytes, of which neutrophils are the most abundant) and endothelial cells [EUROPEAN JOURNAL OF ANAESTHESIOLOGY; Gourdin,MJ; 26(7):537-547 (2009)]. Activated leucocytes and endothelial cells secrete adhesion molecules causing the leucocytes to stick to blood vessel walls, and infiltrate into the ischemic tissue between 6 to 12 hours after reperfusion [CLINICAL NEUROSCIENCE; Kuroda,S; 4(4):199-212 (1997)].
Within and hour of reperfusion, leucocytes adhering to blood vessel walls can completely occlude the vessels, leading to a "no reflow" phenomenon [STROKE; del Zoppo,GJ; 22(10):1276-1283 (1991)], an effect which can be considerably reduced by the monoclonal antibody IB4, which inhibits neutrophil adhesion to the endothelium [STROKE; Mori,E; 23(5):712-718 (1992)]. Swelling of endothelial cells also contributes to the "no reflow" phenomenon. Within a half-hour of ischemia, rat microvessel diameter was reduced to 80% and after an hour was further reduced to 76% of the original diameter [AMERICAN JOURNAL OF PATHOLOGY; Garcia,JH; 145(3):728-740 (1994)].
Permeability of brain capillaries (the Blood-Brain Barrier, BBB) increases progressively in ischemia and reperfusion, ultimately leading to brain edema. BBB permeability to the amino acid leucine increases within 15 minutes of reperfusion injury following an hour of ischemia [STROKE; Sage,JI; 15(1):46-50 (1984)]. Superoxide, nitric oxide, and glutamate interact to contribute to the early stages of BBB permeability increase [NEUROSCIENCE; Kim,GW; 105(4):1007-1018 (2001), PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Bucci,M; 102(3):904-908 (2005), and STROKE; Mayhan,WG; 27(5):965-970 (1996)]. Arachidonic acid also increases BBB permeability, but not in the absence of lipid peroxidation [ADVANCES IN NEUROLOGY; Villacara,A; 52:195-201 (1990)]. Antioxidant enzyme pre-treatment substantially reduces edema in the early stages of reperfusion [STROKE; Armstead,WM; 23(5):755-762 (1992)]. In the inflammatory stage of ischemia/reperfusion injury, the cytokine TNF−α requires the presence of ICAM−1 (InterCellular Adhesion Molecule−1) to increase vascular permeability [AMERICAN JOURNAL OF PHYSIOLOGY; Sumagin,R; 295(3):H969-H977 (2008)].
Edema following reperfusion has been shown to increase for ischemic times up to 3 hours, but not beyond 3 hours [STROKE; Slivka,A; 26(6):1061-1066 (1995)]. Rats subjected to ischemia without reperfusion showed progressively greater brain edema due to sodium entry across the BBB after 12 hours, but albumin did not begin crossing the BBB until after 2 days of ischemia [STROKE; Gotoh,O; 16(1):101-109 (1985)]. From 3 to 48 hours following reperfusion there is an increase in Matrix MetalloProteinases (MMPs) which attack capillary walls to increase permeability and edema [STROKE; Rosenberg,GA; 29(10):2189-2195 (1998)]. MMP inhibitors have been used to protect the BBB against ischemia/reperfusion injury [EXPERIMENTAL NEUROLOGY; Lu,A; 210(2):549-559 (2008), BMC NEUROSCIENCE; Machado,LS; 7:56 (2006), and CHINESE MEDICAL JOURNAL; Zhi-jie,HE; 122(19):2346-2351 (2009)], including statins [BRAIN RESEARCH; Shahanzadeh,AP; 1042(1):1-5 (2005), EUROPEAN JOURNAL OF NEUROSCIENCE; Trinkl,A; 24(2):520-526 (2006), and JOURNAL OF CEREBRAL BLOOD FLOW & METABOLISM; Liu,XS; 26(6):787-796 (2006)].
Treatments to oppose edema formation have also included antagonism of VEGF (Vagal Endothelial Growth Factor, also known as vascular permeability factor) [JOURNAL OF CLINICAL INVESTIGATION; van Bruggen,N; 104(11):1613-1620 (1999)], AVP (Argenine VasoPressin) antagonism [STROKE; Shauib,A; 33(12):3033-3037 (2002), erythropoietin [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Gunnarson,E; 106(5):1602-1607 (2009)], hyperbaric oxygen [BRAIN RESEARCH; Veltkamp,R; 1076(1):231-237 (2006), and tPA (tissue-type Plasminogen Activator) inhibition [BLOOD; Yepes,M; 96(2):569-576 (2000)]. Although tPA can increase perfusion by its thrombolytic action, it can also erode the BBB and be cytotoxic [TRENDS IN NEUROSCIENCE; Yepes,M; 32(1):48-55 (2008)]. A variety of other agents have been used to protect the BBB [NEUROLOGICAL RESEARCH; Takahashi,M; 26(8):862-869 (2004)].
There is a widespread myth that neurons die after a few minutes of ischemia. It would be more accurate to say that neurons subjected to reperfusion injury after a few minutes of ischemia are started on a path that ultimately leads to cell death, but that process can take many hours. In 1976, after dogs were subjected to 12 minutes of cardiac arrest, their circulation was restored using elevated blood pressure, norepinephrine, heparin, and hemodilution with dextran 40. The dogs suffered no neurological damage [ARCHIVES OF NEUROLOGY; Safar,P; 33(2):91-95 (1976)]. This experiment illustrates that neurons are not irreversibly destoyed by 4 to 6 minutes of ischemia, and that resistance to recirculation, not neuron viability, is the cause of the 6 minute limit under normal conditions. In another study, only 36% of neurons were dying after one hour of ischemia followed by two hours of reperfusion [EXPERIMENTAL NEUROLOGY; Li,D; 206(2):280-287 (2007)]. In rats not subjected to reperfusion injury, only 15% of neurons were necrotic after 6 hours of normothermic ischemia [STROKE; Garcia,JH; 26(4):636-642 (1995)].
The myth of neuron death after a few minutes of ischemia is based on the fact that people do not survive if circulation is not restarted within 4 to 6 minutes of cardiac arrest [ AMERICAN JOURNAL OF EMERGENCY MEDICINE; Cummins,RO; 3(2):114-119 (1985)]. Ultimate survival is not the same as immediate death, however. And the ultimate survival of unhealthy human adults who experience cardiac arrest under uncontrolled conditions is an end-point loaded with confounding variables. 46% of mice subjected to 3 minutes of cardiac arrest survived at least 72 hours after reperfusion, while 40% died in the first 24 hours [SHOCK; Menzebach,A; 33(2):189-196 (2010)]. A study of nearly seventeen thousand out-of-hospital human cardiac arrest patients found that only about one in 500 patients were living after one month if the delay between a call and ambulance arrival was between 8 and 12 minutes [HEART; Herlitz,J; 90(10):1114-1118 (2004)].
Calcium (Ca2+) influx into neurons due to the glutamate excitotoxicity of ischemia/reperfusion can be blocked with nimodipine [INTERNATIONAL JOURNAL OF DEVELOPMENTAL NEUROSCIENCE; Babu,CS; 29(1):93-105 (2011)]. TNF−α-receptor blockers can protect neurons from ischemic apoptosis [CELL DEATH & DIFFERENTIATION; Martin-Villalba,A; 8(7):679-686 (2001)], and anti-apoptotic proteins can protect cells against ischemia/reperfusion injury [HUMAN GENE THERAPY; Sawitzki,B; 13(12):1495-1504 (2002)].
Cooling is the most powerful weapon against ischemic damage. Patients resuscitated from cardiac arrest exhibit substantial neurological improvement when subjected to a few degrees of hypothermia [NEW ENGLAND JOURNAL OF MEDICINE; 346(8):549-556 (2002)]. In general, each 10ºC reduction in temperature between 0ºC and 40ºC reduces metabolic rate by one-third to one-half [REJUVENATION RESEARCH; Best,BP; 11(2):493-503 (2008)], although there are discontinuities associated with chilling sensitivity and other specific temperature-dependent activation energies [JOURNAL OF BIOENERGETICS; Raison,JK; 4(1):285-309 (1973)].
The first few degrees of cooling reduces cerebral tissue inflammatory cytokines to one-third the normothermic value, with further reduction by the use of anaesthetic [CRITICAL CARE; Meybohm,P; 14(1):R21 (2010)]. Hypothermia also protects the BBB by depressing activity of proteases [STROKE; Hamann,GF; 35(3):764-769 (2004)]. A combination of buspirone and meperidine can depress the temperature at which shivering could increase metabolism [STROKE; Sessler,PI; 40(11):e614-e621 (2009)]. Rats subjected to 2 hours of cerebral ischemia shared neuroprotective benefits from hypothermia even when begun up to 4 hours after reperfusion [NEUROSCIENCE RESEARCH; Ohta,H; 57(3):424-433 (2007)].
Ischemia and reperfusion are probably more damaging to blood vessels than to neurons, which is positive for the prospects of future reconstruction. Nonetheless, extensive ischemic damage is likely more irreversible than freezing damage in terms of potential repair by future nanomedicine. Ischemically damaged blood vessels become leaky and impede circulation during the perfusion process, blocking cryoprotectant from reaching many areas of the brain to prevent ice formation.
Most ischemic damage to blood vessels in cryonics patients is probably due to free radicals and subsequent inflammatory cytokines, which make antioxidant/spin-trapping agents the most promising protective therapy, along with hypothermia. Good planning by a potential cryonics patient should stress all means to minimize ischemic damage, such as moving near a cryonics facility when terminal, devising alarm systems that warn of cardiac arrest, and attempting to ensure a cryonics standby team is present when cardiac arrest occurs.
![[GO TO BEN BEST'S HOME PAGE]](../homeback.gif) HOME PAGE
HOME PAGE